

‘Junk’ DNA: Evolutionary Discards or God’s Tools?
Originally published in Journal of Creation 14, no 2 (August 2000): 18-30.
* Terms marked with an asterisk are defined in the glossary at the end of the article.
The last decade of the 20th century has seen an explosion in research into the structure and function of the DNA in genomes of a wide range of organisms. As of April 2000, the whole genomes, or full DNA complements of over 600 organisms have been sequenced or mapped.1
The sequence of the fruit fly genome, just completed, has over 130 million base pairs (bp) and is the largest genome sequenced so far.2 The first complete human chromosome has been sequenced,3 and the Human Genome Project expects to complete its work sometime in 2003 [see Human Genome Project Complete ... Again], as does the Mouse Genome Project. Researchers in the new field of genomics-the comparison of the structures, functions and hypothetical evolutionary relationships of the world’s life-forms-are working furiously to deal with the huge inflow of data. Now more than ever, scientists can see at the most basic level the similarities and differences of organisms, and are seeking to understand how the blueprints of cells are decoded and regulated.
A major goal of genomic studies is to understand the role, if any, of the various classes of so-called ‘junk’ DNA. Junk or ‘selfish’ DNA is believed to be largely parasitic in nature, persisting in the genomes of higher organisms as ‘evolutionary remnants’ by their ability to reproduce and spread themselves, or perhaps because they have supposedly mutated into a function the cell can use.
Origin of the junk DNA hypothesis
The idea that a large portion of the genomes of eukaryotes*4 is made up of useless evolutionary remnants comes from the problem known as the ‘c-value paradox’, ‘c’ meaning the haploid* chromosomal DNA content. There is an extraordinary degree of variation in genome size between different eukaryotes, which does not correlate with organismal complexity or the numbers of genes that code for proteins. For instance, the newt Triturus cristatus has around six times as much DNA as humans, who have about 7.5 times as much as the pufferfish Fugu rubripes.5 The c-value between different frog species can differ by as much as 100-fold.6 Early DNA-RNA hybridisation* studies and recent genome sequencing results have confirmed that ›90% of the DNA of vertebrates does not code for a product. Much of this variation is due to non-coding (i.e. not producing an RNA or protein product), often very simple, repeated sequences. With the discovery that many of these sequences seemed to have arisen from mobile DNAs which are able to reproduce themselves, the selfish or parasitic DNA hypothesis was born.7, 8 This said that these sequences served no function in the host organism, but were simply carried on the genome by their ability to replicate or spread copies of themselves within and even between genomes.
Plasterk stated it this way when he wrote about transposons*, one of the ‘junk’ DNA types:
‘This ability to replicate is a sufficient raison d’etre for transposons; they have the same reason for living as, say, the readership of Cell: none. They exist not because they are good, pretty, or intelligent, but because they survive.’ 9
Just as Plasterk was wrong about our reason for living, he is wrong about the purposes of these DNA sequences. Recent research has begun to show that many of these useless-looking sequences do have a function, and that they may have played a role in intrabaraminic10 (‘within-kind’) diversification.
Types of junk DNA
There are four major kinds of junk DNA:
- introns, internal segments in genes that are removed at the RNA level;
- pseudogenes, genes inactivated by an insertion or deletion;
- satellite sequences, tandem arrays of short repeats; and
- interspersed repeats, which are longer repetitive sequences mostly derived from mobile DNA elements.
Introns
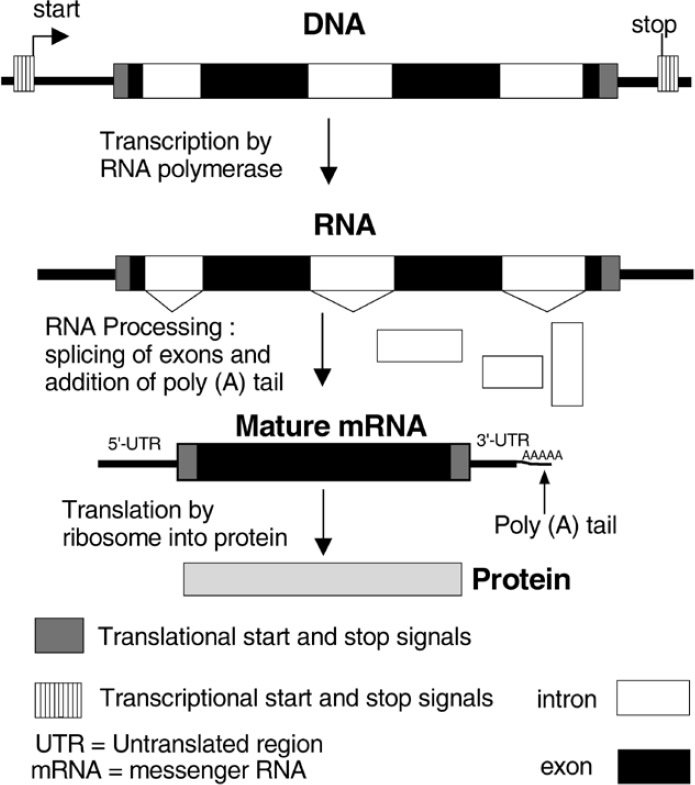
Figure 1. Only portions of a eukaryotic gene code for a protein product.
After most eukaryotic genes and a very few prokaryotic* (bacterial) genes are transcribed, or copied into RNA, there are segments that are cut out of the messenger RNA (mRNA) before it is used as a template to make a protein (Figure 1). Introns in fact form the majority of the sequence of most genes, as was seen when human chromosome 22 was sequenced (Table 1). Why are these RNA pieces present if they are only to be discarded? Evolutionary theory tries to explain these as vestigial sequences, or that they are useful only as sites at which recombination can safely take place to reshuffle exons (coding or protein making segments) into new proteins or new forms of these proteins. Their ubiquity in eukaryotes argues that they are not post-Fall aberrations, but designed features.
What then, could these throwaway segments be doing? There are several possibilities emerging from recent research. One general regulatory role may be to slow down the rate of translation*, as the splicing* process does take time. Alternative splicing allows greater diversity, as certain exons can be skipped and spliced out to allow a different protein to be made from the same mRNA, as is seen in some viruses and in the generation of diversity in antibodies. Another example is the CD6 gene, which is involved in T cell stimulation. Variable splicing of exons gives rise to at least five different forms of the protein, which allows regulation of its activity.11
Another observed mechanism by which introns can regulate gene activity is through the binding of the snipped-out intron RNA to DNA or RNA. There are now a few examples of the role of introns in regulating the genes they are in, as well as other genes. One interesting example is the lin-4 gene intron from the nematode Caenorhabditis elegans. A developmental control gene was found to reside in the intron of another gene (Figure 2).12,13 The small RNA encoded by lin-4 binds to the mRNA of another developmental gene, lin-14, blocking its ability to make protein. The binding site in lin-14 was in another supposedly useless stretch of RNA, the 3’ untranslated region (3'UTR*) found after the last coding region. It was later found that lin-4 RNA also binds to the 3'UTR in another gene in the developmental pathway, lin-28.14 In fact, more and more cases of 3'UTRs performing gene regulatory activities have been observed.15,16
There are examples of protein-encoding genes within introns of other genes that have been recently discovered. For example, on human chromosome 22, the 61-kilobase (kb) TIMP3 gene, which is involved in macular degeneration, lies within a 268-kb intron of the large SYN3 gene, and the 8.5-kb HCF2 gene lies within a 27.5-kb intron of the PIK4CA gene.3
| DNA type | Amount of type (kb) | % of DNA sequenced |
|---|---|---|
| Exons | 390 | 1.40 |
| Introns, UTRs, etc. | 12,406 | 37.00 |
| Pseudogenes | 204 | 0.60 |
| Total Genic region | 13,000 | 39.00 |
| Alu | 5,622 | 16.80 |
| HERV | 161 | 0.48 |
| Line1 | 3,257 | 9.73 |
| Line2 | 1,274 | 3.81 |
| LTR | 256 | 0.77 |
| Other1 | 3151 | 9.42 |
| Total Int. Repeats | 13,721 | 41.01 |
| Short tandem2 | 202 | 0.60 |
| Other tandem | 102 | 0.30 |
| Total Tandem repeats | 304 | 0.90 |
| Total repeats3 | 14,025 | 41.91 |
| Total DNA sequenced | 33,400 | 100.00 |
Some introns also play a role in mRNA editing, a process where the A (adenine) residues in the mRNA are changed to G (guanine).17 Self-complementary* or exon-complementary intron sequences, can bind to each other to form a hairpin loop structure, allowing the sequence of the RNA to be changed after transcription* from the DNA. Thus introns can cause new messages to arise from a gene without altering its DNA coding sequences.
The most general function of introns may be to stabilize closed chromatin* structures in, and around, genes and their associated regulatory DNA elements.18,19 An isochore* is an approximately 300-kb segment of DNA whose base pair composition is uniform above a 3-kb level, for example 67% A-T bp.20 The general ability of an isochore to be transcribed is dependent on the accessibility of its DNA, i.e. how tightly histones* and other DNA-binding proteins wrap up the DNA. This is seen as being at least partially dependent on the A-T or G-C bp content of a segment of DNA. Though this content can be skewed somewhat by the choice of triplet codons* used in the coding DNA (since the code is redundant21), exons are still constrained in their ability to vary the bp content. The presence of introns throughout genes allows the proper levels to be maintained, and indeed introns reflect the general isochore type much more closely than the coding regions. The presence of introns may well be a condition for at least some forms of sectorial repression like superrepression, where large sections of chromatin are altered to turn off groups of cell-type-specific genes or developmental genes. It was shown, for example, that the gene for rat growth hormone, when deprived of its introns, was no longer able to form its normal more condensed structure when reinserted back into cells.22
It is important to know whether the specific sequence of an intron is required for its function when constructing phylogenetic or family trees, or when determining baraminic* placement of an organism. In evolutionary studies, DNA sequence comparisons are used to try to build phylogenetic trees to trace ancestors to descendants. Since introns are generally believed to be free from the constraints of functionality when mutations cause changes in their sequence, introns in a particular gene are often compared between organisms, with the bp differences seen between their sequences supposedly indicating the degree and time of divergence since they last shared a common ancestor. In some instances, the assumption that an intron is likely to have mutated freely and extensively during the presumed millions of years of evolutionary history has proved wrong. Koop and Hood found that the DNA of the T cell receptor complex, a crucial immune system protein, is 71% identical between humans and mice over a stretch of 98-kb of DNA. This was an unexpected finding, as only 6% of the region encodes protein, while the rest consists of introns and non-coding regions around the gene.23 Does it follow then that we have a recent common ancestor with mice? Since this does not fit in with evolutionary theory, the authors conclude instead that the region must have specific functions that place constraints on the fixation of mutations. This illustrates that DNA sequence comparisons to establish evolutionary relationships are not the independent tests that they are claimed to be. If the data do not support the desired evolutionary theory, ad hoc explanations of altered rates of mutation, functional constraints, etc., can be brought in to explain away discrepancies.24
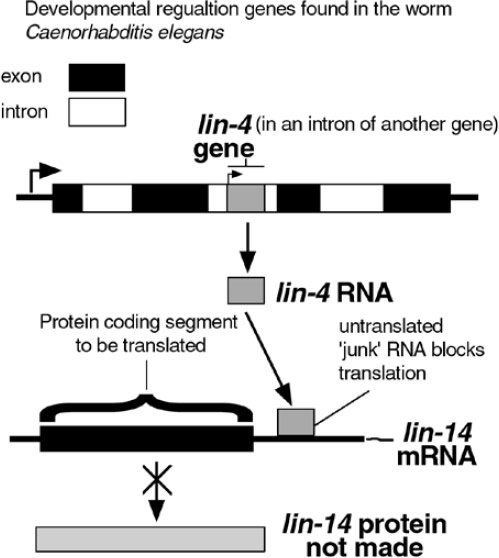
Figure 2. Interaction of two ‘junk’ RNAs regulates a developmental gene.
Another example of selective interpretation of DNA sequence comparison data using introns is the study of an intron in an important sperm maturation gene on the Y chromosome of humans.25,26 It was hoped that the ancestry of modern humans could be traced by sequencing this 729-bp intron from 38 different men from different ethnic groups. Surprisingly, all 38 men had exactly the same sequence, which was then interpreted as a ‘recent’ common ancestor (27,000-270,000 years ago) for the whole human race, or possibly that the intron had functional constraints on its mutability. This latter premise was rejected by the authors because the sequence of the same intron in chimp, gorilla and orangutan was progressively more different. These data would strongly support the biblical creationist view that there was a severe bottleneck in the human population when the Flood reduced the varieties of Y chromosomes to the one shared by Noah and his sons. Apes would not be expected to have exactly the same sequence as humans, as they are from separate created kind(s). The fact that they do have a similar intron argues for a function for this sequence, and the intron may have been originally created slightly different for proper function in an ape versus a human.
Thus evidence is mounting to support the important role of introns in gene regulation and chromosome structure, which would remove 8.15%27 of the junk DNA of the human genome from the trash heap.
Pseudogenes*
Occasionally located near functional genes or gene families, there are sequences that very closely resemble other functional genes, but have been inactivated in someway. Some have a mobile element inserted in their open reading frames (ORFs*), others seem to be ‘processed’ genes, i.e. they look as though the RNA from another gene has been reverse transcribed (RNA used as a template to make DNA) and reinserted back into the DNA (Figure 3). A processed pseudogene* thus precisely lacks the introns, possesses 3'-terminal poly-(A) tracts*, and lacks the upstream promoter* sequence required for transcription of the corresponding parent gene. Pseudogenes are common in mammals, but virtually absent in Drosphila.28 Nineteen percent of the coding sequences identified in human chromosome 22 were designated as pseudogenes, because they had significant similarity to known genes or proteins but had disrupted protein coding reading frames. 82% appeared to be processed pseudogenes.3 Many pseudogenes have additional mutations in them, presumably because there is no functional constraint on their mutation. For example, the human beta-tubulin gene family consists of 15-20 members, of which five have these pseudogene hallmarks.29 Some pseudogenes affect gene activity by binding transcriptional factors that activate the normal gene. Whether this is intentional design or something the organism has simply adjusted to is difficult to say. Many pseudogenes do seem to fit the profile of true junk DNAs.
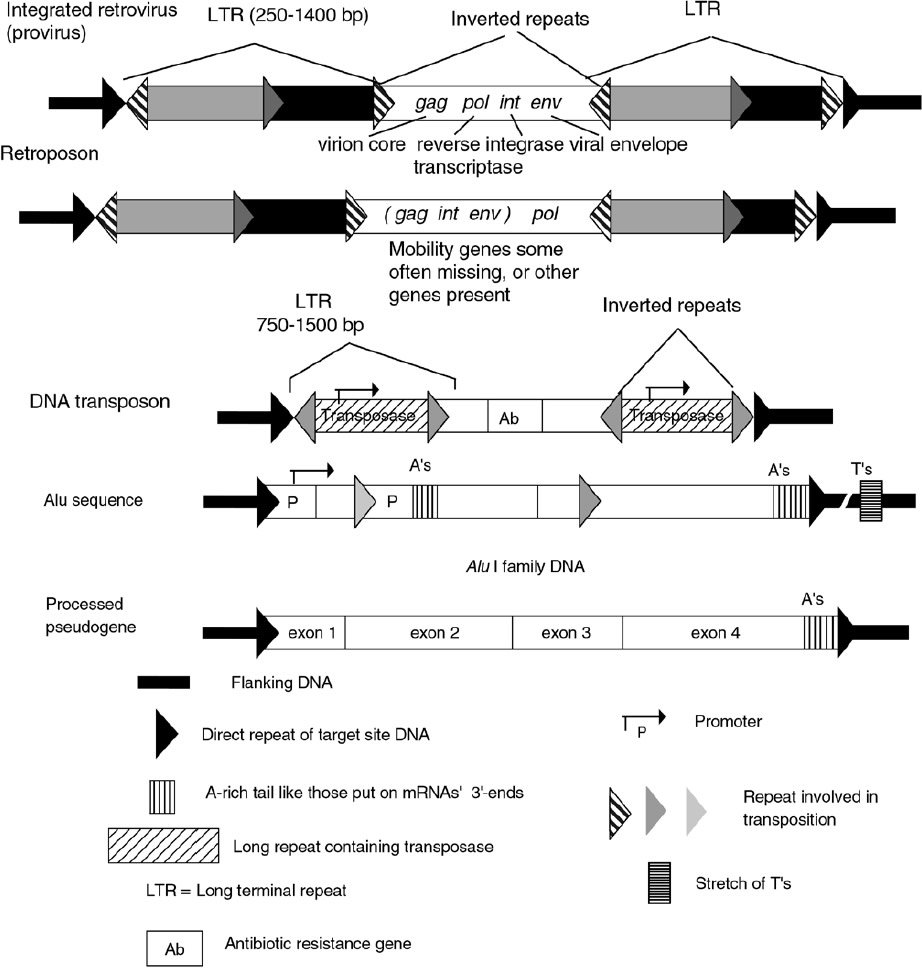
Figure 3. Comparison of integrated mobile DNA sequence structures.
Repetitive DNA sequences, including mobile DNA sequences
Repetitive DNA sequences form a substantial fraction of the genomes of many eukaryotes (Table 1, Table 2).30,31 This class includes satellite DNA (very highly repetitive, tandemly repeated sequences), minisatellite and microsatellite sequences (moderately repetitive, tandemly repeated sequences), the new megasatellites (moderately repetitive, tandem repeats of larger size) and transposable or mobile elements (moderately repetitive, dispersed sequences that can move from site to site; see Table 2).
When first discovered, they did not seem to confer any benefit to the host organism, as their ability to move about the genome and/or cause recombination between different homologous copies has often resulted in deleterious mutation and disease. We now know that at least some of these sequences carry out important functions.
Satellite sequences
The functionality of a sequence of 2 or 3 bp repeated a thousand or so times is not immediately apparent. In addition, the lengths and compositions of these repetitions often vary wildly between species, between organisms of the same species, or even between cells of the same organism. But greater understanding has come as scientists realize how DNA acts not only as the information source for the cell, but also as the library in which it is housed.32 It is beginning to be seen that the dispensability of sequences is not an indicator of their non-functionality, and that in many cases, repetitive sequences tend to fill functions collectively rather than individually.
Satellite sequences vary in their repeat size and in their array size (Table 2). Microsatellites are the smallest, at a repeat size of as little as 2 bp, and the newly discovered megasatellite sequences, which actually can contain ORFs, are 4-10 kb long.33 The actual sequence repeated differs from species to species, and repeats can differ slightly from one another. The number in an array can vary between individuals, which is why forensic DNA fingerprinting techniques use mini- and microsatellite differences to identify individuals.
| Sequence types | Repeat size (bp) | Array size (kb) | Copy Number1 | Functions, features of family members |
|---|---|---|---|---|
| Satellites — large tandem arrays | 10–25% of total DNA | |||
| Microsatellite | 2–5 | 0.2–0.5 | 3 x 105 | Repeat expansion causes cancer |
| Minisatellite | ~15 | 0.5–3 | 105 | Changes in sequence cause cancer |
| Satellite | 5–100 | 100,000 | 107 | Centromere and telomere function |
| Megasatellite | 4–10 kb | 30–100 | 30–100 | ? |
| Interspersed elements | 35–40% of total DNA | |||
| Retrotransposons | ||||
| LTR-containing elements | ||||
| copia2, gypsy2 | ~5 kb | NA | 20–60 |
Can befound as free circular DNA Horizontal transfer of genes; can infect germline cells |
| Yeast Ty | 6.3 kb | NA | 40 | Ty1 and Ty3 transpose specifically to genes transcribed by RNA polymerase III; Repair of chromosomal breaks |
| Poly-A elements | ||||
| LINE1 (L1) | 1–7 kb | NA | ~105 |
Mutant sequences can promotecancer Some provide polyadenylation signals Some copies mobile |
| HeT-A, TART2 | 6–10 kb | 5–10 | ~106 | Maintenance of telomeres |
| SINEs | ||||
| Alu | 300 | NA | ~106 |
Retinoic acid receptor-binding site Enhancer of gene activity Silencer of gene activity Negative calcium response element Alters protein synthesis Insertion can cause disease |
| Retroviruses | ~6–10 kb | NA | Infections capability | |
| HIV-1 | Can cause disease, cancer, AIDS | |||
| HERVs3 | Provide polyadenylation signals | |||
| DNA transposons | ||||
| P element2 | 2.9 kb | NA | 10–1002 | Horizontal transfer of genes, speciation; germline-specific transposition |
| Mariner/Tc1 | 1.5 kb | NA | ~107 | Horizontal transfer of genes |
| MITEs | 125–500 bp | NA | 103–104 | Associated preferentially with genes in plants; can provide regulatory sequences |
Functions of satellite sequences
The first recognised function of these types of sequences was in organising the centromeres, the constricted sites on each chromosome where the chromosomes attach to cellular tethers and are pulled apart during meiosis and mitosis. These sequences help condense the DNA region they are in into heterochromatin*.
One hypothesis of the collective functionality of repeat sequences is that long stretches of noncoding sequences act as tethers, permitting placement of groups of genes into different zones in the cell nucleus.19 Transcriptionally inactive heterochromatin and the heterochromatin-like telomeric sequences (sequences at the end of chromosomes), may associate their respective chromatin segments much of the time with the nuclear periphery. Very long runs of gene-poor, AT-rich isochores*, would be the tethers that permit the gene-rich, GC-rich isochores to distribute themselves into the appropriate nuclear zones for transcription and RNA processing.
The importance of the sequences of satellite DNA is reflected when these sequences are mutated. A mutation in a minisatellite just after the end of the Harvey ras gene (which encodes a growth regulatory protein) may contribute to as many as 10% of all cases of breast, colo-rectal and bladder cancer, and acute leukemia. The mutant minisatellites bind a transcriptional regulatory factor,34 which causes an abnormal increase in transcription of the Harvey ras gene (Figure 4).
Retroviruses* and retroelements*
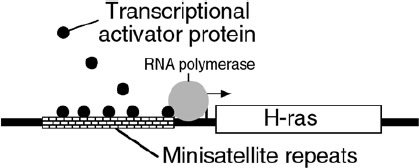
Figure 4. Mutations in minisatellite DNA can result in cancer. Mutated satellite DNA near the Harvey ras gene (a major regulator of cell growth) can bind a protein that increases ras activity.
These ‘class I mobile elements’ reproduce themselves through an RNA intermediate which, in a reversal of the usual DNA to RNA transcription, is reverse transcribed to DNA by the reverse transcriptase* enzyme encoded on intact elements. One of the remarkable findings of the human genome project is that a high percentage (35.40%) of human nuclear DNA consists of dispersed retroelements (Table 1).35 Short and long interspersed elements, SINEs and LINEs, make up the majority of this class of DNA, with Alu and LINE-1 (L1), respectively, being most abundant in humans.36 L1 elements encode their own reverse transcriptase, that probably is also responsible for the spread of SINEs, which lack this enzyme. HIV-1, the AIDS virus, human endogenous retroviruses* (HERVs), and solitary long terminal repeats (LTRs*) apparently derived from HERVs, are also part of this class of retroelements (Figure 3, Table 2).
Most eukaryotic retrotransposons* move only sporadically in the genome. An exception is the hybrid dysgenesis seen in Drosophila, where if flies containing a retrotransposon are mated to flies not containing the particular retrotransposon, the element transposes with a high frequency, resulting in death or mutation of many of the progeny. Host factors, many not well characterized as yet, seem to keep the transposition rate in check (see below).
Functions of retroelements
Do these abundant elements have functions, or have hapless eukaryotic genomes been parasitized by selfish DNA? There are more and more examples of these elements performing important functions. One example is the Alu family. This 300-bp sequence (named for the enzyme used to identify it) occurs almost a million times in the human genome, up to 3.5% of the total DNA (Table 2). It is estimated, and has been seen in many cloned genes, that there are 4 or 5 Alu elements in every gene. Despite their number, they have been generally considered parasitic DNA, with occasional deleterious effects on the genome when they exercise their ability to retrotranspose to sites in and near genes, or recombine with each other abnormally. Such disruptions have caused neurofibromatosis, or elephant man’s disease.16 Mutations in the Alu sequence also have been associated with cancer. Alu sequences have been found to affect the functions of at least 8 different genes (Table 2).37,38 Though Alu sequences do have internal promoters for RNA polymerase III (an enzyme which transcribes genes encoding RNAs needed for translation of mRNA into protein), normally very little RNA is produced from all these Alu sequences. However, under certain stressful conditions such as a viral infection, these transcripts increase dramatically and affect protein synthesis levels to help the cell deal with the stress.39 Thus, though individual Alu elements have a very weak effect, hundreds of thousands of them together can affect protein synthesis.
Epigenetic control mechanisms, or modifications of gene activity that are due to modifications of the DNA itself and not its sequence (see below), are associated with repeats. A repeat-induced process involving L1 retroelements has been hypothesised for X-chromosome inactivation, which is necessary to maintain proper gene dosage in females, who have two X chromosomes (Table 2).40
Endogenous retroviruses (that is, those that are obtained from inheritance rather than infection) can also affect gene expression. The LTRs of two such viruses provides the sequence signal for the polyadenylation* of the mRNA of two newly discovered human genes.41 An L1 repeat was found to provide the polyadenylation signal for the mouse thymidylate synthase gene. Retrotransposons were also seen to help in repairing chromosomal breaks in yeast. Retroelements modulate expression of many more genes.42
DNA transposons*
DNA transposons, or ‘class II transposable elements’, move from place to place by replicative transposition (that increases the copy number) or by a simple cut-and-paste mechanism. Though in general not as common or in as high a copy number as retroelements, they are still found in most organisms. Examples are the Drosophila P elements, bacterial transposons such as Tn10 and Tn7, the Mu phage, and the ubiquitous mariner/Tc1 superfamily of transposons. The mariner/Tc1 family is the most widespread, being found in most insects, flatworms, nematodes, arthropods, ciliated protozoa, fungi and many vertebrates, including zebra fish, trout and humans.43 Copy number varies from two copies in Drosophila sechellia, to 17,000 in the horn fly Haematobiairritans, accounting for 1% of the genome. The vast majority of them appear to have been inactivated by multiple mutations. The close homology between mariner/Tc1 elements found in species thought to have diverged 200 million years ago has fuelled the hypothesis that these elements can transfer horizontally (that is, not by normal inheritance) between different species, or even different phyla (see below). Again, the evolutionist gets to pick and choose from his smorgasbord of explanations when the data do not fit the evolutionary tree.
Miniature inverted-repeat transposable elements (MITEs)
A recently discovered third class of mobile elements is the miniature inverted-repeat transposable elements (MITEs).44,45,46 They are very small (125-500 bp), and have short terminal inverted repeats. They were first found in plants, but have also been found in nematodes, humans, mosquitoes and zebrafish.47,48,49,50 They are found in the thousands and tens of thousands per genome, and have been given colourful names (e.g. Tourist, Stowaway, Alien and Bigfoot) to reflect their apparent ability to move about in the genome. Their mechanism of transposition is still unknown, but they appear to be DNA elements that cannot move about on their own (non-autonomous). Though none seem to be presently active, they are believed to have been mobile in the recent past because of the high levels of sequence similarity between elements in a particular family, and the differences in insertion sites seen in closely related species.51 MITEs are particularly interesting in terms of generating genetic variation in that they are preferentially associated with genes (see below).46,52
Effects of mobile and repetitive elements on gene expression
Mobile elements and repetitive elements can alter the structure and regulate expression of the genome in several different ways. As described earlier, transposition can disrupt genes by direct insertional mutagenesis and can adversely affect transcription. Many retrotransposons have strong constitutive (always on) promoters that can cause inappropriate expression of downstream genes. If the promoter is in the opposite direction of the gene, RNA complementary to the mRNA of the gene can be made that can act as antisense RNA* that binds up the mRNA, affecting translation.
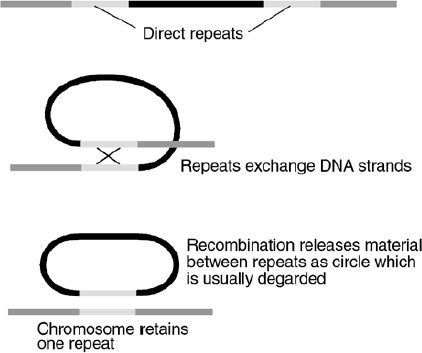
Figure 5. Recombination between direct repeats causes the loss of the DNA between them.
Recombination between similar DNA strands is a necessary process for repair of DNA breaks and allele* shuffling between homologous chromosomes. But the presence of mobile and repetitive elements in inappropriate positions can result in recombination products that are deleterious, such as translocations*, inversions*, and other chromosomal rearrangements (Figure 5). For example, it was shown that a widespread chromosomal inversion commonly seen in Drosophila buzzatii is caused by the recombination between two copies of a transposable element in opposite orientations.53 There can even be an exchange of DNA between non-homologous chromosomes: such as was seen in maize, in this case mediated by the recombination of one complete and one partial copy of the Ac (Activator) transposable element.54
Target site selection in mobile DNA
Many of the retrotransposons and DNA transposons seem to have very little site-specificity in where they integrate.55 Integration sites for most mammalian and Drosophila retroelements appear to be distributed more or less randomly in the genome. Vertebrate retroviruses do have a general preference for insertion into regions with an open chromatin configuration.56
However, there are some specific ones that do show target selectivity.51 R2 is a non-LTR retrotransposon that inserts preferentially in the 28S ribosomal RNA genes of various insect species. Group II introns present in some yeast mitochondrial genes (genes carried in the energy-producing organelles in the cell), are mobile elements very similar to poly (A)-type retrotransposons. After copying themselves, they can reinsert precisely back into their spots between two exons. Their ability to move argues for their spread into various genes at some point in time. The yeast retrotransposons Tyl and Ty3, integrate preferentially upstream of genes transcribed by RNA polymerase III, which transcribes genes needed for protein synthesis.
Very recently, evidence has been found that certain P elements* containing regulatory sequences from developmental genes, showed a high frequency of reinserting at the parent gene (homing) and preferential insertion at another site containing regulatory genes.57
The first example known of a host using the movement of a retrotransposon to its advantage, was found in the telomere maintenance of Drosophila. The telomeres, or chromosomal ends of Drosophila, are maintained differently than any other known organism. Two retroposons, HeTA and TART, are present in multiple copies on the telomeres, and will retropose specifically to the end of the telomere and heal a frayed chromosome.58
Observed regulation of mobile DNA
Epigenetic mechanisms, or reversible but heritable changes in chromatin structure, are seen to play a role in regulating genes. Methylation of cytosine residues, modification of the DNA-binding histones, and production of antisense RNAs, are some of the mechanisms by which gene expression can be modified without permanent genetic change to the gene regulated.59 Methylation of the cytosine residues of DNA is used by the cell to turn off genes not currently needed. Cytosine methylation inactivates the promoters of most viruses and transposons (including retroviruses and Alu elements). In fact, transposons are so abundant, rich in CpG dinucleotides and heavily methylated, that we now know that the large majority of 5'-methylcytosine in the genome actually lies within these elements.60 This prevents the movement of the elements under normal circumstances. Thus transposable elements that integrate into promoters of genes can alter gene expression patterns by attracting methylation or chromatin modifications to regulate the modified promoter.53
Drosophila, in general, are very vulnerable to mutation by mobile element activity. From 50-85% of all spontaneous mutations seen in the fruit fly are due to transposon insertions.53 But Drosophila does have one type of host control in the recently identified gene named flamenco. Flamenco normally acts to keep the gypsy retrotransposon in check. When flamenco is mutated, gypsy transposes at a high frequency in germ line (reproductive) cells.50
Criteria for identifying junk DNA
There are several possible scenarios for the presence and function of the putative junk DNA sequences described above:
- They all perform God-designed functions in present day organisms in their present form and location, though current research has not revealed what those are as yet. This is unlikely, as it seems clear that in some individuals and species, the placement or particular sequence of one of a family of non-coding DNAs can lead to deleterious effects such as cancer and genetic disease. This would contradict the Bible’s description of God’s original perfect creation.
- All non-coding sequences could have been created with functions, but some have lost their functions due to God’s purposeful limitations, and/or accumulation of mutations post-Fall. This would fit in with our observation of the rest of creation, where, though the perfection of God’s design can be seen, it has become obscured by consequences of the Fall, allowing death and suffering to enter the world.
- There is the possibility that some of the elements, such as the mobile elements in particular, have never had designed functions. Rather, they are pieces of degenerate DNA affected by the Fall that randomly move about and mutate genomes, causing only deleterious effects.
The ability of DNA sequences to rearrange and/or to move about in the genome or even between genomes, was originally a heretical idea for both evolutionist and creationist, but now is one that is strongly supported as being an integral part of gene regulation. Many systems utilizing similar recombination and rearrangement mechanisms are necessary for important cellular functions, such as the process of DNA repair, rearrangement of DNA segments to form the genes for the thousands of different antibodies, the yeast mating type switching system, the flagellar switching system of Salmonella, and the antigen switching system of the malaria parasite. Therefore, the second scenario seems the most likely.
A working list of criteria needs to be developed to attempt to identify DNA sequences that may actually fit the category of junk DNA. The presence of some junk DNA would be expected due to the fallen state of genomes. True junk DNA may have one or more of the following characteristics:
- The DNA element is present within another gene, insertionally inactivating it.
- The DNA element is not found at that location in other members within the same species.
- The effects of the presence of the element, if known, are deleterious, e.g. lead to cancer, genetic disease, etc.
- The element can be deleted without any observed ill effects on the organism or many generations of its descendants.
- The sequence of the element closely matches that of a mobile element, or contains a mobile element sequence.
For example, pseudogenes have many of these junk DNA characteristics, though their transformation into junk DNA may in some cases have been intentionally arranged by God for the purpose of rapid diversification of created kinds.
The AGEing theory and diversification
There are, as described above, instances of functions for transposable DNAs, but until recently there has not been a particular purpose ascribed to repetitive and mobile elements as a group. A new hypothesis formulated by genomicist and creationist Wood addresses the past and present functions of mobile and repetitive DNA.61
Since these elements are capable of rapid change of the genome, and can even be transmitted horizontally between species, he proposes that God designed them to move about or recombine in the genomes of organisms to allow the rapid intrabaraminic diversification seen in the 500 years or so after the Flood. He sees their role as being designed to act for a limited period of time, after which they would be inactivated by mutation or repression by other regulatory elements. He proposes that such elements should be renamed Altruistic Genetic Elements (AGEs) to emphasize that their purpose is different than that proposed for ‘selfish’ DNA.
The AGEs are hypothesised to work by activating dormant genes or inactivating active genes, or by horizontally transferring genetic information between species or possibly baramins with AGEs in the form of mobile elements. The phenotypic changes would be primarily cosmetic, such as variations in size or coloration, or would involve activation of a complex of genes needed to utilize a new environmental niche, like the Arctic fox’s adaptation to cold. There is a need for creationists to explain how a holobaramin such as the cat family,62 could diversify into the many species of cats that were present even in Job’s time in just a few thousand years or possibly a few hundred years. Currently observed genetic mechanisms and natural selection are far too slow to explain this rapid speciation. A limited time period of AGE activity could explain how this rapid diversification could occur.
If, for example, the proposed AGEs were at work in the diversification of the equines, we have the testable predication that differences in size, morphology and coloration could be traced back to the genetic level by mobile or repetitive DNA elements located near genes controlling coloration. Pseudogenes and relic retroviral sequences could then be the result of the action of an AGE gone wrong after its designed activity began to fail. The AGEing theory could also solve the ‘founding pair' problem-that is, when a rare macromutation occurs in an individual such that it cannot successfully hybridise with its parental species, this mutation is lost unless it can mate with another animal with the same mutation.
For this proposed AGEing process to work, at least three things must be observed in putative AGEs:
- They must show site specificity in where they insert, or evidence that they had such specificity in the past.
- Transmission of AGEs between organisms horizontally and into germline DNA is required.
- We should see AGEs associated with genes affecting size, morphology, coloration, and specialised environmental adaptation rather than housekeeping genes.
As for the first requirement, though many mobile elements are not specific in their target sites, there are examples of those that are, as discussed above. Since AGE movement is supposed to have occurred largely in the past, we might expect to see only a few with the intact capability.
As for the second requirement, horizontal transmission*, the evidence for that occurring has become very strong,63 and in the case of the P and gypsy elements in Drosophila, such transmission has actually been observed occurring between species. Originally, no wild-caught D. melanogaster contained the P element and laboratory stocks collected 60 years ago reflected this. Then gradually, more and more wild-caught flies contained the element originally found in D. willistoni, until now all wild flies even in remote locations contain this element.64 Recently, it was also shown that the copia retrotransposon from D. melanogaster was transferred to D. willistoni (probably via a parasitic mite).65,66 There was also a report that gypsy-free fruit flies permissive for transposition of the gypsy retroposon could incorporate gypsy into their germline DNA when larvae were fed on extract of infected pupae.67 There is no obvious evidence pointing to a functional change mediated by these horizontal transfers, but the principle is there.
As for the third requirement, are there any examples known now of mobile or repetitive elements that can cause these types of phenotypic changes? In bacteria, there are many examples of transfers of antibiotic resistance mediated by transposons,68 and the horizontal transfer of genes, though in general prokaryotes have comparatively little 'junk’ DNA. Some evolutionary researchers now propose that mobile elements may be involved in speciation. Mobility of a retroelement was activated in a cross between two wallaby species, though the hybridisation resulted in only sterile males.69 In maize, the original studies of Nobel Prize winner Barbara McClintock demonstrated that the activity of the transposons in different corn kernel cells could be followed by their effects on corn kernel coloration. In plants, there is additional strong evidence that movement of mobile elements in the past has altered gene expression. Although retrotransposon sequences, for example, are seldom found near genes in animals, recent analyses of plant mobile element insertion sites have revealed the presence of degenerate retrotransposon insertions adjacent to many normal plant genes that act as regulatory elements.70 In addition to retrotransposons, MITEs are also found adjacent to many plant genes, where they also often provide regulatory sequences necessary for transcription.71 Plants, as well as animals, would have had to adjust to the drastically-altered post-Flood world. Other, more dramatic examples may exist, and further research will hopefully reveal them.
AGEing activity during the Ark sojourn
Putting thousands of animals aboard the Ark could have had a dual purpose. Not only did it preserve their lives, but also it would probably allow transfer of genetic material and/or activation of latent genes simultaneously in all land animals. Under the relatively crowded Ark conditions, transfer of genetic material from one species to another through broad host range viruses, parasitic mites or fleas would be facilitated. This might have produced a distribution of AGEs in species in such a way as to defy evolutionary phylogeny, as is seen for the Tc1/mariner family and in the gypsy family of retrotransposons.72
Why debunk ‘junk’ DNA?
What is the relevance to creation science, and to Christians in general, of a better understanding of the function of these DNA elements? Because of the publicity surrounding the Human Genome Project, there is increasing general interest in how our genomes work, and what exactly they look like. There is more and more emphasis being placed on discovering our evolutionary history through DNA, not fossils.
The fact that functions are being found for junk DNAs fits in well with creation science, but was not predicted by evolutionary theory, though of course the theory is being adjusted again to accommodate the data. The intricate flexibility and specificity of these ‘junk’ DNA sequences are a strong testimony to a Creator who plans and provides for the future of his creation.
Glossary
Allele-one of several alternate forms of a gene occupying a given locus on a chromosome. Return to text.
Antisense RNA-RNA made by copying the other DNA strand in a coding segment in the opposite direction; this RNA will bind to the mRNA made from the coding or sense strand.Return to text.
Baramin-the creationist term for an original created kind as described in Genesis; not synonymous with species. Organisms within the same baramin may be of different species but can cross-hybridise, like the horse and the donkey. Return to text.
Complementary-two strands of DNA or RNA are said to be complementary when they can form base pairs (A-T, G-C) with each other, e.g. AATTCC and TTAAGG. Return to text.
Chromatin-the complex of DNA and protein in the nucleus of the interphase cell. Return to text.
Euchromatin-the less condensed chromatin in the nucleus that is more transcriptionally active than the heterochromatin. Return to text.
Eukaryote-an organism with an organized nucleus. Return to text.
Haploid-half the set of the chromosome pairs; contains one copy of each chromosome pair and one of the sex chromosomes; characteristic of gametes (sperm and egg cells). Return to text.
Heterochromatin-regions of the genome that are in a highly condensed state and are not usually transcribed. Constitutive hetereochromatin is always in this condensed, inactive state, contains no genes, and is usually found at the centromeres and teleomeres. Facultative heterochromatin is condensed only in certain cell types, or at certain developmental stages when the genes contained in it need to be turned off. Return to text.
Histones-a family of basic proteins found tightly associated with DNA in all eukaryotic nuclei; their binding forms a bead structure called a nucleosome. Return to text.
Horizontal transmission-when mobile elements or viruses are transferred between individuals by infection rather than by inheritance (vertical transmission). Return to text.
Human endogenous retroviruses (HERVs)-retroviruses that have become part of the human genome in the past by insertion into the germline cells. Return to text.
Hybridisation-the pairing of single-stranded complementary RNA and/or DNA strands to give an RNA-DNA or DNA-DNA hybrid. Return to text.
Inversion-occurs when recombination between DNA segments causes the DNA between them to be flipped into the opposite orientation at the same chromosomal locus. Return to text.
Isochore-an approximately 300 kb segment of DNA whose bp composition is uniform above a 3 kb level, for example 67% A-T bp. This is believed to enable a certain level of co-regulation of all the DNA in the isochore. Return to text.
LTR-long terminal repeat; the longer, more complex repeated sequences at the ends of some mobile elements, which are required for them to transpose. Return to text.
ORF-open reading frame; a stretch of DNA or RNA that contains of series of triplet codons coding for amino acids, without any protein termination codons, that is potentially translatable into protein. Return to text.
P elements-DNA transposons found in fruit fly species that often have a high level of mobility. Return to text.
Promoter-a region of DNA involved in binding of RNA polymerase to initiate transcription. Return to text.
Poly-(A) tail-a sequence of adenine residues added to the 3’ end of a mRNA after transcription in the process called polyadenylation; believed to help stabilize mRNAs from being degraded. Return to text.
Pseudogene-a gene that has been inactivated in the past by an insertion or deletion of DNA. Return to text.
Prokaryote-an organism that lacks an organized nucleus, and has its DNA mostly in a single molecule; a bacterium. Return to text.
Processed pseudogene-a gene that has been apparently reverse-transcribed from its mRNA back into DNA and reinserted into a chromosome. It thus lacks its introns, has a poly-A tail, and often is bounded by the characteristic direct repeats associated with transposition. Return to text.
Retroelement-any sequence that transposes through an RNA intermediate. Return to text.
Retrotransposons-mobile elements that encode reverse transcriptase. Transpose through an RNA intermediate. Classed into LTR-containing and poly (A)- containing:
LTR-containing-similar to proviral form of vertebrate retroviruses and usually have 2 ORFs, gag and pol (protease, integrase, reverse transcriptase, RNase H), e.g. Gypsy and tom.
Poly (A)-containing retroelements or retroposons lack LTRs and have a 3’ A-rich region. Have 2 ORFs, gag and pol. Some elements such as L1 and the I Factor of Drosophila, contain a reverse transcriptase. L1 is found in yeast and humans. Return to text.
Retrovirus-a virus using RNA as its information storage system rather than DNA, integrates into host DNA as part of its lifecycle in a way very similar to retrotransposons, but also has additional genes that code for its packaging into virus particles for infection of other hosts. Return to text.
Reverse transcriptase-an enzyme found in retroelements that will make a complementary DNA strand from an RNA template. Return to text.
Splicing-two exons, or coding regions on a messenger RNA, are joined together when the intron (non-coding segment) between them is removed. Return to text.
Translation-the synthesis of protein on the messenger RNA template. Return to text.
Translocation-of a chromosome describes a rearrangement in which part of a chromosome is detached by breakage and then becomes attached to some other chromosome. Return to text.
Transcription-synthesis of RNA on the DNA template. Return to text.
Transposase-the enzyme that cuts the target DNA and splices in the transposing sequence; called the integrase in retroelements.
Transposon-any DNA sequence that can move about the genome, either by replicating itself, or by a cut-and-paste mechanism. In its simplest form, it is a transposase gene (see above) surrounded by a sequence on either side repeated directly or in inverse form, e.g. ATTGCGC and CGCGTTA are inverted repeats. Return to text.
Triplet codon-three nucleotides in an RNA or DNA that signal the insertion of a particular amino acid or termination signal; e.g. AUG would be the ‘code word’ for methionine. Return to text.
UTR-untranslated region; the parts of a messenger RNA before the first exon ('5 prime’ UTR) and after the last exon ('3 prime’ UTR) that are not translated into protein (non-coding). Return to text.
Acknowledgements
The author wishes to thank Dr. Todd C. Wood for providing unpublished information on his AGEing theory and his rice genome research. Thanks also to the editor for helpful discussions, information and patience with revisions.
Linda K. Walkup has a B.A. in biochemistry and biology from Rice University, and a Ph.D. in molecular genetics from the University of New Mexico Medical School. She is actively involved in the New Mexico Creation Science Fellowship, teaches creation science classes and gives seminars on molecular biology-related creation/evolution issues. She is currently employed in homeschooling her two children and organizing the local homeschooling science fair. Return to top.
Footnotes
- National Centre for Biotechnology Information Website, <www.ncbi.nlm.nlh.gov>.
- Adams, M.D., et al., Celniker, S.E, Holt, R.A, Evans, C.A., et al., . . . and Venter, J.C., The genome sequence of Drosophila melanogaster, Science 287(5461):2185–2195, 2000.
- Dunham, I., et al., The DNA sequence of human chromosome 22, Nature 402(6761):489–495, 1999.
- Terms marked with an asterisk are defined in the Glossary at the end of the article.
- Brenner, S., Elgar, G., Sandford, R., Macrae, A., Venkatesh, B. and Aparicio, S., Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome, Nature 366(6452):265–268, 1993.
- Lewin, B., Genes, John Wiley and Sons, NY, p. 294, 1985.
- Doolittle, W.F. and Sapienza, C., Selfish genes, the phenotype paradigm and genome evolution, Nature 284(5757):601–603, 1980.
- Orgel, L.E. and Crick, F.H.C., Selfish DNA: the ultimate parasite, Nature 284(5757):604–607, 1980.
- Plasterk, R.H.A., Molecular mechanisms of transposition and its control, Cell 74(5):781–786, 1993.
- Baramin is a creationist term meaning ‘created kind’. See Wise, K.P., Practical baraminology, CEN Tech. J. 6(2):122–137, 1992.
- Bowen, M.A., Whitney, G.S., Neubauer, M., Starling, G.C., Palmer, D., Zhang, J., Nowak, N.J., Shows, T.B. and Aruffo, A., Structure and chromosomal location of the human CD6 gene: detection of five human CD6 isoforms, J. Immunol. 158(3):1149–1156, 1997.
- Lee R.C., Feinbaum, R.L. and Ambros, V., The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14, Cell 75(5):855–862, 1993.
- Wightman, B., Ha, I. and Ruvkun, G., Post-transcriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans, Cell 75(5):843–854, 1993.
- Moss, E.G., Lee, R.C. and Ambros, V., The cold shock domain protein LIN28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA, Cell 88(5):637–646, 1997.
- Wickens, M. and Takayama, K., Deviants — or emissaries, Nature 367(6458):17–18, 1994.
- Nowak, R., Mining treasures from ‘junk DNA’, Science 263(5147):608– 610, 1994.
- Herbert, A., RNA editing, introns and evolution, Trends Genet. 12(1):6–9, 1996.
- Zuckerkandl, E. and Hennig, W., Tracking heterochromatin, Chromosoma 104(1–2):75–83, 1995.
- Zuckerkandl, E., Junk DNA and sectorial gene repression, Gene 205(1–2):323–343, 1997.
- Bernardi, G., The human genome: organization and evolutionary history, Ann. Rev. Genet. 29:445–475, 1995.
- Each amino acid attached to a transfer RNA can be added onto a protein when the transfer RNA anticodon recognizes the complementary three nucleotides (a ‘word’) in the mRNA. Since there are only 20 amino acids and there are 64 possible trinucleotide combinations or ‘words’, several triplet codons can code for the insertion of one amino acid. This redundancy allows some variation in G-C content in a gene without changing the composition of the protein produced.
- Liu, K., Sandgre, E.P., Palmiter, R.D. and Stein, A., Rat growth hormone gene introns stimulate nucleosome alignment in vitro and in transgenic mice, Proc. Nat. Acad. Sci. USA 92(17):7724–7728, 1995.
- Koop, B.F. and Hood, L., Striking sequence similarity over almost 100 kilobases of human and mouse T-cell receptor DNA, Nature Genet. 7(1):48–53, 1994.
- Jerlström, P., Shaky tree of life, CEN Tech. J. 13(1):10–11, 1999.
- Batten, D., Junk DNA (again), CEN Tech. J. 12(1):5, 1998.
- Dorit, R.L., Akashi, H. and Gilbert, W., Absence of polymorphisms at the ZFY locus on the human Y chromsome, Science 268(5214):1183–1185, 1995.
- Duret, L., Mouchiroud, D. and Gautier, C., Statistical analysis of vertebrate sequences reveals that long genes are scarce in GC-65 isochores, J. Mol. Evol. 40(3):308–317, 1995.
- Petrov, D.A., Lozovskaya, E.R. and Hartl, D.L., High intrinsic rate of DNA loss in Drosophila, Nature 384(6607):346–349, 1996.
- Watson, J.D. and Hopkins, N.H., Roberts, J.W., Steiz, J.A. and Weiner, A.M., Molecular Biology of the Gene, The Benjamin/Cummings Publishing Company, Inc., Menlo Park, CA, p. 662, 1987.
- Cavalier-Smith, T. (ed.), The Evolution of Genome Size, John Wiley, Chichester, 1985.
- John, B. and Miklos, G.L.G., The Eukaryote Genome in Development and Evolution, Aleen and Unwin, London, 1988.
- Bodnar, J., Telephone book of life, Nature 361(6413):580, 1991.
- Gondo, Y., Okada, T., Matsuyama, N., Saitoh, Y., Yanagisawa, Y. and Ikeda, J.E., Human megasatellite DNA RS447: copy-number polymorphisms and interspecies conservation, Genomics 54(1):39–49, 1998.
- Krontiris, TG., Minisatellites and human disease, Science 269(5231):1682– 1683, 1995.
- Jurka, J., Repeats in genomic DNA: Mining and meaning, Curr. Opin. Struct. Biol. 8(3):333–337, 1998.
- Smit, A.F.A., The origin of interspersed repeats in the human genome, Curr. Opin. Genet. Dev. 6(6):743–748, 1996.
- Britten, R.J., DNA sequence insertion and evolutionary variation in gene regulation, Proc. Nat. Acad. Sci. USA 93(18):9374–9377, 1996.
- Britten, R.J., Mobile elements inserted in the distant past have taken on important functions, Gene 205(1–2):177–182, 1997.
- Chu, W-M., Ballard, R., Carpick, B.W., Williams, B.R.G. and Schmid, C.W., Potential Alu function: regulation of the activity of double-stranded RNA-activated kinase PKR, Mol. Cell. Biol. 18(1):58–68, 1998.
- Neumann, B., Kubicka, P. and Barlow, D.P., Characteristics of imprinted genes, Nature Genet. 9(1):12–13, 1995.
- Mager, D.L., Hunter, D.G., Schertzer, M. and Freeman, J.D., Endogenous retroviruses provide the primary polyadenylation signal for two new human genes (HHLA2 and HHLA3), Genomics 59(3):255–263, 1999.
- Tomilin, N.V., Control of genes by mammalian retroposons, Int. Rev. Cytol. 186:1–48, 1999.
- Hartl, D.L., Lohe, A.R. and Lozovskaya, E.R., Modern thoughts on an ancyent marinere: function, evolution, regulation, Ann. Rev. Gen. 31:337–358, 1997.
- Bureau, T.E. and Wessler, S.R., Tourist: a large family of small inverted repeat elements frequently associated with maize genes, Plant Cell 4(10):1283–1294, 1992.
- Bureau, T.E. and Wessler, S.R., Mobile inverted-repeat elements of the Tourist family are associated with the genes of many cereal grasses, Proc. Nat. Acad. Sci. USA 91(4):1411–1415, 1994.
- Bureau, T.E. and Wessler, S.R., Stowaway: a new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants, Plant Cell 6(6):907–916, 1994.
- Oosumi, T., Garlick, B. and Belknap, W.R., Identification of putative nonautonomous transposable elements associated with several transposon families in Caenorhabditis elegans, Mol. Evol. 43(1):11–18, 1996.
- Smit, A.F. and Riggs, A.D., Tiggers and DNA transposon fossils in the human genome, Proc. Nat. Acad. Sci. USA 93(4):1443–1448, 1996.
- Tu, Z., Three novel families of miniature inverted-repeat transposable elements are associated with genes of the yellow fever mosquito, Aedesaegypti, Proc. Nat. Acad. Sci. USA 94(14):7475–80, 1997.
- Izsvak, Z., Ivics, Z., Shimoda, N., Mohn, D., Okamoto, H. and Hackett, P.B., Short inverted-repeat transposable elements in teleost fish and implications for a mechanism of their amplification, J. Mol. Evol. 48(1):13–21, 1999.
- Zhang Q., Arbuckle, J. and Wessler, S.R., Recent, extensive, and preferential insertion of members of the miniature inverted-repeat transposable element family Heartbreaker into genic regions in maize, Proc. Nat. Acad. Sci. USA 97(3):1160–1165, 2000.
- Mao, L., Wood, T.C., Yu, Y., Budiman, M.A., Woo, S., Sasinowski, M., Goff, S., Dean, R.A. and Wing, R.A., A survey of rice (Oryza sativa) transposable elements from a complete library of STC sequences, Genome Research, in press.
- Caceres, M., Ranz, J.M., Barbadilla, A., Long, M. and Ruiz, A., Generation of a widespread Drosophila inversion by a transposable element, Science 285(5426):415–418, 1999.
- Zhang, J. and Peterson, T., Genome rearrangements by nonlinear transposons in maize, Genetics 153(3):1403–1410, 1999.
- Craig, N.L., Target site selection in transposition, Ann. Rev. Biochem. 66:437–474, 1997.
- Labrador, M. and Corces, V.G., Transposable element-host interactions: regulation of insertion and excision, Annu. Rev. Genet. 31:381–404, 1997.
- Taillebourg, E. and Dura, J.M., A novel mechanism for P element homing in Drosophila, Proc. Nat. Acad. Sci. USA 96(12):6856–6861, 1999.
- Pardue, M.L., Danilevskaya, O.N., Lowenhaupt, K.Y., Slot, F. and Traverse, K.L., Drosophila telomeres: new views on chromosome evolution, Trends Genet. 12(2):48–51, 1996.
- Wolffe, A.P. and Matzke, M.A., Epigenetics: regulation through repression, Science 286(5439):481–486, 1999.
- Yoder, J.A., Walsh, C.P. and Bestor, T.H., Cytosine methylation and the ecology of intragenomic parasites, Trends Genet. 13(8):335–340, 1997.
- Wood, T.C., The AGEing process: post-Flood intrabaraminic diversification caused by altruistic genetic elements (AGEs), Baraminology Conference 99, unpublished paper.
- Robinson, D.A. and Cavanaugh, D.P., Evidence for the holobaraminic origin of the cats, Creation Res. Sci. Quart. 35(1):2–14,1998.
- Syvanen, M., Horizontal gene transfer: evidences and possible consequences, Annu. Rev. Genet. 28:237–261, 1994.
- Daniels, S.B., Peterson, K.R., Strausbaugh, L.D., Kidwell, M.G. and Chovnick, A., Evidence for horizontal transmission of the P-tranposable element between Drosophila species, Genetics 124(2):339–355, 1990.
- Jordan, I.K., Matuyunina, L.V. and McDonald, J.F., Evidence for the recent horizontal transfer of a long terminal repeat retrotransposon, Proc. Nat. Acad. Sci. USA 96(22):12621–12625, 1999.
- Houck, M.A., Clark, J.B., Peterson, K.R. and Kidwell, M.G., Possible horizontal transfer of Drosophila genes by the mite Proctolaelaps regalis, Science 253(5024):1125–1129, 1991.
- Kim, A., Terzian, C., Santamaria, P., Pelisson, A., Purd’homme, N. and Bucheton, A., Retroviruses in invertebrates: the gypsy retrotransposon is apparently an infectious retrovirus of Drosophila melanogaster, Proc. Nat. Acad. Sci. USA 91(4):1285–1289, 1994.
- Hall, R.M., Mobile gene cassettes and integrons: moving antibiotic resistance genes ingram-negative bacteria, CIBA Found. Symp. 207:192–202, 1997.
- Jerlström, P., Jumping wallaby genes and post-Flood speciation, CEN Tech. J. 14(1):9–10, 2000.
- Labrador and Corces, Ref. 51, p. 397.
- Bureau, T.E., Ronald, P.C. and Wessler, S.R., A computer-based systematic survey reveals the predominance of small inverted-repeat elements in wild-type rice genes, Proc. Nat. Acad. Sci. USA 93(16):8524–8529, 1996.
- Miller, K., Lynch, C., Martin, J., Herniou, E. and Tristem, M., Identification of multiple gypsy LTR-retrotransposon lineages in vertebrate genomes, J. Mol. Evol. 49(3):358–366, 1999.
Support the creation/gospel message by donating or getting involved!

Answers in Genesis is an apologetics ministry, dedicated to helping Christians defend their faith and proclaim the good news of Jesus Christ.
- Customer Service 800.778.3390
- Available Monday–Friday | 9 AM–5 PM ET
- © 2026 Answers in Genesis