

Are Pseudogenes ‘Shared Mistakes’ Between Primate Genomes?
Originally published in Journal of Creation 14, no 3 (December 2000): 55-71.
Abstract
The claim that pseudogenes and their respective variations are shared between primates in a nested hierarchy, and can only be explained through common evolutionary descent, is found wanting.
Summary
‘Given a sufficient lack of comprehension, anything (and that includes a quartet of Mozart) can be declared to be junk. The junk DNA concept has exercised such a hold over a large part of the community of molecular biologists …(emphasis in original).’ – Zuckerkandl and Henning1
‘DNA not known to be coding for proteins or functional RNAs, especially pseudogenes, are now at times referred to in publications simply as nonfunctional DNA, as though their nonfunctionality were an established fact.’ – Zuckerkandl, Latter and Jurka2
The evolutionary claim that pseudogenes and their respective variations are shared between primates in a nested hierarchy, and can only be explained through common evolutionary descent, is found wanting. Evidence for pseudogene function continues to accumulate, and is much more significant than the actual number of known functional pseudogenes. In addition, pseudogene-related phenomena show considerable differences between ‘close’ primates, and are neither self-consistent nor in agreement with other phylogenetic interpretations. Furthermore, pseudogene deployment and alteration are governed by strongly non-random events. Unless evolutionists can rigorously demonstrate that pseudogene-related phenomena cannot occur independently in different primates, their ‘shared mistakes’ argument should be rejected.
The human genome is believed to be littered with pseudogenes, which are gene-like structures that do not code for proteins because of some presumed defect.3 A recently-published4 abridged example is shown (Table 1). Useful summaries on this topic are available.5,6 The term pseudogene, as used here, encompasses both the classical and the retroposited varieties, the latter of which includes interspersed repeats*, notably SINEs* and LINEs*.7 Creationist scientists (including me) generally assume that God would not create purposeless genes in different primates, and that God did not independently disable the same genes in humans and nonhuman primates during the Curse.
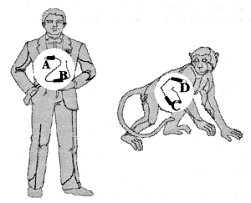
Figure 1.Schematic illustration of orthologs and paralogs. A, B, C and D
represent any combination of mutually-similar and presumably-related genes and/or
pseudogenes. A and B are always paralogs of each other, as are C and D. Depending upon
degree of similarity (and therefore perceived evolutionary relatedness), the following
orthologous pairings are possible: (A, C), (A, D), (B, C) or (B, D). Only the first and
fourth, or second and third, orthologous pairings can simultaneously
coexist.
Unfortunately, the distinction between empirical observation and evolutionary interpretation is often particularly difficult in molecular biology. There is always an element of subjectivity in the process of aligning sequences of homologous (orthologous*) DNA8,9 (Fig. 1), and this is aggravated by non-corresponding segments of the same.10 Furthermore, it is unclear just how close the resemblance must be to rule out a fortuitous match-up of mistakenly orthologous sequences. For instance, there is ambiguity11 about the status of one 34 bp (base-pair*) segment exhibiting 68% nucleotide* correspondence between the human and rat genomes. And last, molecular similarities, including those of pseudogenes, do not create self-evident truths, but must be interpreted:
‘At face value, this is just wrong—alignment procedures delineate similarity between sequences but tell us nothing about their common ancestors, if such ever existed. To give an absurd but relevant example, poly-A* tails of any two processed pseudogenes are perfectly alignable, but it would be a stretch to consider them homologous.’12
Contrary to the assertions of some,6 the presumed temporal persistence of supposedly-useless pseudogenes actually constitutes a serious problem for evolution. The manufacture of DNA is energetically costly to the cell, and natural selection should remove DNA were it actually useless.13 A mechanism for removal is now known.14
If they are actually selectively neutral and subject to random mutations, ‘old’ pseudogenes should in fact be scrambled beyond recognition. Apropos to this, orthologous SINEs have now been found in different phyla,15 and the cited researchers recognize that the (evolutionary) maintenance of a close correspondence between such phylogenetically*-distant organisms is very difficult to explain if SINEs are of no use to their carriers. More on this later.
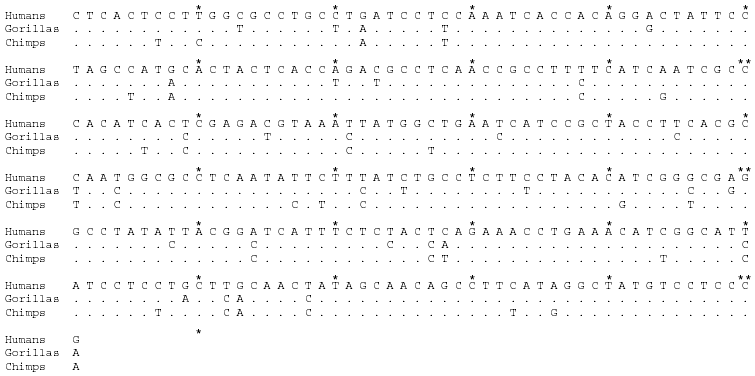
Table 1. Aligned sequences of Cytochrome b and mitochondrial-related
pseudogenes. From Moreira and Seuanez (1999). Base abbreviations are as follows:
A-adenine, C-cytosine, G-guanine and T-thymine. The 301-member sequence is demarcated by
tens (*) and hundreds (**). Nucleotides identical to human are denoted
(.).
Click thumbnail to view the entire table.
I. Are pseudogenes useless?
If pseudogenes are functional, they are no different from any other homologous structure found in nature. These all reflect the fact that God used the same ‘blueprint’ or ‘art form’ repeatedly when constructing different living things. In this case, the orthologous placement of pseudogenes, and their respective differences, are moot.
The importance of pseudogene-caused genetic diseases6 is apt to be exaggerated because, by their very nature, deleterious retroelements* are so obvious.16 The opposite is the case with beneficial pseudogenes. In fact, for at least some pseudogenes, failure to observe them coding a product under experimental conditions is not ipso facto proof of their inability to do so:
‘In these and other examples it cannot be stated with certainty that a gene is unequivocally either a pseudogene or a gene. It is possible that analysis has not been performed in the appropriate temporospatial conditions to detect expression.’17
One argument adduced in support of pseudogene nonfunction is the observation that they contain many more nucleotide differences (which are assumed to be mutations), and are more variable in terms of base-pair composition than their paralogous* protein-coding genes. Yet this observation is compatible with function.2 In fact, as mentioned below, an ability to code for a product useful to the host organism hardly exhausts the possibilities for pseudogene function. It is interesting to note that the inferred nucleotide-substitution rate in pseudogenes shows only crude correspondence with primate phylogeny, for which reason it has to be manipulated post hoc by up to tenfold18–20 in order to contrive an agreement between the timing of different episodes of primate evolution. Pseudogenes whose age is deduced on the basis of the numbers of nucleotide differences from their coding paralogs show only a weak relationship between age and the numbers of indels*.21 Each branch of the phylogenetic tree, of pseudogenes relative to primate evolution, exhibits widely divergent rates of indel formation.22
It is interesting to note that there are some pseudogenes which cannot be straightforwardly portrayed as inactivated copies of their paralogous genes. This includes the human AS pseudogenes, each of which shares a concerted pattern of 19 nucleotides that sets it apart from its inferred gene paralog.23
A large and rapidly-growing body of evidence for pseudogene functionality exists, most of which will be presented in a forthcoming paper.24 Earlier-known evidences are given elsewhere.5 There is a theory25 which proposes that pseudogenes interact with antisense RNA*. The functionality of Alu* units has long been suspected,26 and recently confirmed.27,28
The distinction between ‘processed genes’ and ‘processed pseudogenes’ is not, contrary to one critic,6 the result of creationist confusion, but is instead the product of the critic’s semantics. After all, the former is but a functional version of the presumably-nonfunctional latter.29 Evolutionists assume that certain retropseudogenes have become ‘recruited’ by evolutionary processes and are thereby secondarily functional. These are called processed genes. But this of course begs the question about them having lost function to begin with! The claim6 that functioning pseudogenes are manifestations of beneficial mutations is also an egregious act of begging the question. The latter also reflects the following prejudicial and erroneous notion: if ‘crippling mutations’6 prevent a protein-coding function, this is ipso facto synonymous with no function.
A numbers game? Depreciating evidences of pseudogene function
Max’s response6 to this evidence is to use the ‘ATT’ (Appeal to Technicalities) fallacy and the ‘ATM’ (Appeal to Marginalization) fallacy.30 Each is described in turn.
Users of the ATT fallacy engage in much post hoc quibbling about the broad applicability of contrary evidence.31 One example32 is the belittling of the discovery of ‘junk DNA’ function33 by pointing out the (correct) fact that this noncoding DNA differs from that in pseudogenes. But discoveries of this nature, and successive ones,34 cannot be dichotomized so easily (see Abstract). This is especially so in light of the fact that the identical ‘nonfunctional unless proven functional’ mentality besets our understanding of all types of noncoding DNA. Finally, and as noted earlier (and especially in the forthcoming paper24), evidence for function is not limited to generic ‘junk DNA’, but is now known for representatives of all the major types of pseudogenes. Therefore, attempts to depreciate the significance of such function (as by asserting that it is only true of a few processed pseudogenes6) appears to be another use of the ATT fallacy.
The ATM fallacy treats evidence as a simple numbers game.35 But, as pointed out by the philosopher of science Sir Karl Popper,36 evidence cannot be treated in this way (e.g. as so many points for, versus so many points against, a theory). Indeed, one contrary observation is often sufficient to falsify a theory. Popper’s philosophy clarifies the fact that, contrary to Max,6 the ‘nonfunctional pseudogenes’ argument is not substantiated by large numbers of apparently nonfunctional pseudogenes, but is instead falsified by a significant and rapidly growing body of evidence which demonstrates such function.
II. The overall nonfunction of pseudogenes: established or tentative?
In response to those who assume that pseudogene nonfunction is well established,6 we must consider several factors, not the least of which is the following:
‘Science is supposed to advance step by step, with all conclusions supported by adequate evidence. Yet conclusions are sometimes widely accepted without much evidence, and woe to those who come along later with data supporting what is already “received” wisdom.’37
Apropos to this, it is acknowledged38,39 that nonfunctional-pseudogene beliefs took hold at a time when the genome was little understood, and when sociobiology dominated all of biology,40 favouring such attitudes. The classic essays by Orgel, Crick, Doolittle and Sapienza, which largely inspired the notion that noncoding DNA is useless, parasitic and ‘selfish’, are recognizably anthropomorphic and speculative.41 In addition, Howard and Sakamoto40 stress the fact that, majority opinion notwithstanding, pseudogene-nonfunction beliefs rest largely upon negative evidence. In stark contrast to Max,6 others are not so sure that we know, even to this day, what pseudogenes cannot do (see also Abstract):
‘Short interspersed repetitive DNA elements (SINEs) are found in various eukaryotes* … . We still do not know the biological significance of these elements and how these elements evolved to the present status.’42
‘Do these elements [LINEs and SINEs] serve a generally useful function or are they simply “selfish DNA”?’43
‘However, this is not a strong argument, and whether L1* is “selfish” remains to be determined.’ 44
‘The problem is that generally one does not know whether a pseudogene has any noncoding phenotypic effect and whether the effect is deleterious or advantageous.’45
In addition, the ‘few known functional pseudogenes implies few functional pseudogenes’ thinking, though presented by Max6 as virtual fact, is recognizably no more than a hypothesis.46 Moreover, this hypothesis is either explicitly or implicitly rejected by various investigators, who recognize the fact that the relatively small number of known functional pseudogenes is not at all commensurate with their overall significance:
‘There are severe limits to our recognition of the roles of mobile elements … the knowledge of all of the control elements that may be important to genes is still very restricted. Since mobile elements occur and carry out useful functions in positions many kilobases from the initiation of transcription even those significant mobile elements that have been inserted within the last few million years may not have been principally recognized. Thus it can be argued that 21 examples represent a large number.’47
‘The question then is: which of the hundreds of thousands of Alu inserts are contributing to the regulation of nearby genes, and which are without significant effect?’48
Not surprisingly, the perceived rarity of functional pseudogenes has been self-perpetuating:
‘… given the fact that there are a million Alu elements in the human genome and there have been no systematic studies to identify which of them have regulatory functions, it must be only be a matter of time before human-specific Alus are found to control gene expression (emphasis added).’28
‘Recognizing that Alu repeats might be junk DNA, most researchers chose to study their mobility and incidental effects on genome structure, as opposed to their possible function.’49
Other investigators40 have also discussed how low expectations of pseudogene function have been self-fulfilling.
Apropos to this, it is erroneous to compare overall pseudogene function to a defendant in a criminal trial pleading innocence because evidence favourable to him may emerge in the future.6 To begin with, he is actually seeking acquittal as a result of the current state of evidence.50 Second, evolutionists cite ‘use current evidence only’ arguments6 selectively, i.e. for pseudogenes, but certainly not for naturalistic theories for life’s origins, otherwise they would admit the complete inadequacy of such theories, and acknowledge an external Designer. But a double standard is followed instead, and we are assured that no Designer is needed because, ‘Even though today we cannot explain life’s origins mechanistically, one day we probably will.’
III. Pseudogene deployment: ‘shared mistakes’ between primate genomes?
Do pseudogenes themselves form a nested hierarchy*?
A large fraction of most pseudogenes differ considerably from their paralogous genes. For instance, a compilation of 65 primate pseudogene sequences,51 totalling 80.6 kb*, indicates that parts of the pseudogene sequences resemble their paralogs at not much higher than chance levels (50% for two unrelated strands of DNA). Less than one-third of the 80.6 kb aggregate sequences are 85% similar to their paralogs, and a very small unspecified fraction of the same reaches 90%. The authors point out that progressively lower levels of similarity mean progressively greater ambiguity as to the origins and the timing of the accumulated pseudogene/gene differences. Taken to its logical conclusion, this means that ‘shared mistake’ arguments cannot even have relevance, let alone validity, for a large fraction (perhaps the majority) of pseudogenes.
Numerous pseudogenes consist of multiple paralogous copies in each primate genome. In such cases, ‘shared mistakes’ take on a life of their own. Evolutionists must essentially ‘shop around’ for the closest match52 in trying to deduce the orthologous pairings of pseudogenes from primate to primate. This can also occur in the case of multiple Alu repeats.53 If evolutionary ‘trees’ indicate an anomaly in which the pseudogenes of distantly-related primates resemble each other more closely than those of more closely-related primates, this can always be blamed after-the-fact on either an artefact of the ‘tree’ itself, or on an incorrect pairing of orthologs.54
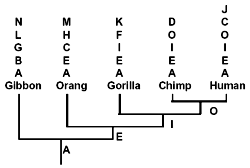
Figure 2. A schematic phylogeny illustrating the hierarchical (vowel) and
non-hierarchical (consonant) deployment of ‘shared mistakes’ among five
primates. These ‘mistakes’ can be either the orthologous pseudogenes
themselves or the variations of one orthologous pseudogene to another, or
both.
Let us now consider those pseudogenes which have only single copies per primate genome. In doing this, I will adhere to the evolutionary methodology of counting only shared similarities and dissimilarities each of which simultaneously differs from that of ‘less derived’ primates.6 Even so, as shown below, while some pseudogenes appear to be hierarchically shared (as illustrated in Fig. 2) between primates,6 others definitely are not. Most of the latter are apomorphic*. (C), however, is an example of phylogenetic discordancy: it occurs in humans and orangs, but not in any primates of intermediate evolutionary derivation.
Years ago, I had called attention to a pseudogene which was shared by humans and gorillas but not chimps.55 It has since been alleged that the chimp pseudogene is lacking because its locus* had been deleted.56 This is an inference which rests on the assumption that all primates are evolutionarily related, and so any differences in DNA sequences must be of secondary origin. Other phylogenetic studies may have ignored missing loci.57 This complication, usually reckoned ‘missing information’, eventually makes any phylogenetic analysis uninformative.58
Moreover, missing loci cannot come to the rescue of evolutionists in still other hierarchy-defying instances of pseudogene deployment:
‘These include two of the OR genes (hOR17-7 and OR17-209), which are intact in human and chimpanzee, but are pseudogenes in gorilla, due to one-base deletions*. In both cases, the gorilla pseudogenes are accompanied by an intact variant, a potential case of heterozygosity with one of the alleles being a pseudogene.’59
Other examples of gorilla-only pseudogenes are given below. Otherwise, one OR pseudogene is human-specific and another set of OR pseudogenes are shared by humans and chimps but, ironically, are believed to be of independent origin.
Evolutionists can always invoke the ‘gene inactivation occurred after divergence’ claim, after the fact, in such situations, but such thinking is admittedly an assumption.60 More pointedly, this ad hoc rationalization begs the question about pseudogenes forming a phylogenetic nested hierarchy in the first place. And it is far from the only one. Gene conversions* can also be invoked for apomorphic pseudogenes, as was the case with the human-only BC200-Beta pseudogene.61
In other primates, the deployment of known pseudogenes also often fails to conform to a nested evolutionary hierarchy. The spider monkey has an apomorphic gamma-globin pseudogene.62 Elsewhere, seemingly orthologous DRB3 pseudogenes in the tamarin and titi contain different ‘inactivating mutations’. According to evolutionary storytelling, once upon a time some genes had come to resemble each other by convergence* before each one of them had become a pseudogene.63 In still another example, we encounter an exact reversal of the usual evolutionary expectation of genes increasingly becoming converted into pseudogenes in progressively more derived primates.6 Apropos to this, an inferred inactivation of the theta-1 globin genes exists in the less-derived non-primates (e.g. rabbit) and in the less-derived galago, but it is the more-derived higher primates that have functional orthologs instead.64 As a final example, nuclear pseudogenes in the primate family Cebidae portray a confusing phylogenetic picture, and this is largely blamed on confounding homoplasies* among the pseudogenes.65
Ironic to those who highlight pseudogenes as an accumulation of ‘shared mistakes’,6 there are evolutionists who are suspicious of pseudogenes as a means of charting the course of primate evolution:
‘Pseudogenes appear to be subject to virtually no selection and have, therefore, been used to provide the missing data. However, most pseudogenes are members of gene families in which frequent exchange of sequences among members may complicate interpretations of sequence divergence and phylogeny.’66
Finally, to put the ‘shared pseudogenes’ argument in a broader context, note that evolutionists cannot even agree as to which particular genomic structures can only be explained by shared evolutionary descent. The mitochrondrial gene order in birds has been shown to arise independently.67 The MHC complex exhibits considerable similarities among primates, with most of these genetic motifs believed to predate the chimp-human divergence.68 Yet, in a major about-face, evolutionists now recognize that complex MHC genetic motifs can arise independently.63,69 They currently reckon only 7 of 13 allelic lineages, and only at most a few of the 135 alleles of the DRB1 locus, as predating the human-chimp split.70
Do nested hierarchies characterize interspersed repeats?
Both creationists and evolutionists recognize the fact that the majority of classical pseudogenes have always been located close to their protein-coding paralogous genes. But retropseudogenes are believed to have been retrotransposited at considerable distances from the paralogous parent gene, and only shared evolutionary ancestry is supposed to be able to account for such coincident placement in different primates.6 The most numerous retropseudogenes, by far, are SINEs (especially Alus71) and LINEs (notably L1 elements), each of which number in the hundreds of thousands72 in the human genome alone. Evolutionists believe that these elements are periodically inserted during the course of primate evolution (Fig. 3), and that each such episode generates a unique new family of interspersed repeats, creating markers suitable for phylogenetic analyses.
There are, however, numerous rationalizations available for dealing with inserted elements that fail to conform to a nested hierarchy. Contrary to the claim that successive families of LINEs and SINEs are hierarchically deployed among animals, there are many instances where clearly intact loci lack the predicted interspersed element. This occurs between members of different species73 as well as different orders.74 The rationalization invoked is this: the LINE or SINE element did not happen to integrate into that part of the host population which eventually survived into the present.
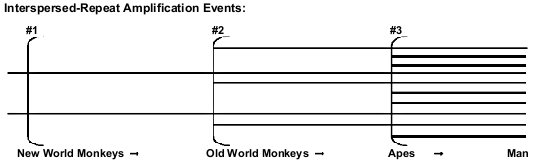
Figure 3.Idealized and schematic portrayal of successive amplifications (#1, #2
and #3) of progressively-younger (thicker-dash) families of SINEs. Between episodes of
retroposition, the source gene(s) supposedly accumulate mutations. This causes each
successive ‘printout’ of retroposited SINEs to differ from previous ones by
up to several unique nucleotide substitutions. These nucleotide differences define a new
SINE family. Similar considerations apply to LINE elements, but many of these elements
can indirectly copy themselves.
Evolutionists have long believed that Alu insertion* is an irreversible process; hence the absence-presence of an Alu at an orthologous site constitutes an ipso facto primitive-derived polarity. Were a formerly-inserted Alu to undergo subsequent deletion, this event would supposedly be betrayed by the simultaneous deletion of some of the flanking sequence*.6 To the contrary, precise excisions of Alu units can occur: a gorilla-human shared Alu is absent at the orthologous chimp locus, and an extra 12 bp right Alu-flanking repeat, added to an empty-site sequence, marks the missing-Alu spot.75
Members of the ‘wrong’ family of inserted repeats can even share particular orthologous sites. In one instance, an old-family Alu in the gibbon was found to be situated at the orthologous site of a new-family Alu in gorillas, chimps and humans.76 The former was then assumed to be a template for the evolution of the latter. In another instance,77 a modern-family Alu unit was found in humans, located anomalously at the site expected for an orthologous older-family unit. So a gene conversion event was conjured up, after the fact, for having supposedly reconfigured and ‘modernized’ the onetime old Alu family member to make it nearly identical to a modern human-specific Alu family member.
For the longest time, many evolutionists have argued that the parallel* insertion of essentially identical retropseudogene units, at the orthologous site in different animals, is a virtual impossibility. One estimate placed the odds against such an event at one in many billions.78 Wouldn’t you know it—the same SINE units,79,80 as well as LINE units,81 have now been discovered independently emplaced at orthologous sites in different genomes.
For the vast majority of the ostensibly-younger Alus, there can be no question about their occurrence in a nested hierarchy, as the vast majority of them are apomorphic.78 Furthermore, the Ya5 Alu family is a showcase of a violated nested hierarchy. Originally believed to occur only in humans, Ya5 Alu repeats turned up in chimps,82 and then gorillas. So it was then supposed that the source gene had generated Ya5 retropseudogenes prior to the human-chimp-gorilla divergence, and so, in accordance with a nested hierarchy, these ape Ya5 Alus would also be found at the orthologous sites in humans. But they were not, and this development was thus explained away:
‘However, it is also remarkable that according to our interpretation, the PV EPL must have been active at least once in each of the three divergent HCG lineages.’76
Remarkable indeed. We are seriously asked to believe that the PV EPL source gene became activated independently in all three primates, and many times in two of them, after their mutual divergence. The plasticity of organic evolution is a sight to behold!
Of course, the belief that families of interspersed elements form nested hierarchies is predicated on the belief that the families are factual entities. But, not only are apomorphic nucleotide substitutions found, but also ones which appear, disappear, and then reappear again in ostensibly progressively more derived Alu families.83 The same occurs in L1 families.74 In addition, there are so many recent L1 families in existence that they have no clear-cut boundaries, and it is admittedly difficult to sort out the resulting ‘inconsistent pattern of shared characters’.84 Such blurring also occurs between the older Alu families.85
Earlier, I noted that the molecular ‘clock’ varies considerably from one pseudogene to another. The same holds for the rate of nucleotide substitutions in Alu units. The accumulation of what may ironically be called unshared-mistake nucleotide differences, between orthologous human-chimp Alu elements, differ significantly from one Alu element to another. Obviously, independent of ‘age’ and degree of evolutionary relatedness, nucleotide-substitution rates turn out to be governed by the base composition of the host DNA.86
How certain is the orthologous pairing of retropseudogenes?
Can we really be sure that the same interspersed repeat is located at the identical location in different primate genomes? Evolutionists commonly believe that orthologous inserted-element units and orthologous flanking sequences (including any flanking repeats) can all be unambiguously identified. The actual situation is not as clear-cut. As discussed below, orthologs are usually far from identical, and there are features which reduce the distinctiveness of each inserted element from another.
To begin with, the Alu units themselves, apart from varying in terms of nucleotide sequence, do not even have to be of equal length to be judged orthologous.78 In particular, the differences in length of the poly-A tail, between presumably orthologous Alu units, are often excused on the basis of the vulnerability of homopolymeric* sequences to episodes of partial deletion after insertion.87 In addition, the direct repeats* which usually surround each retropseudogene often have ambiguous boundaries with the Alu unit itself and/or the surrounding flanking sequence.26 Furthermore, owing to the prevalence of (A+T) upstream of Alu insertions,88 the direct repeats are also (A+T)-rich, thereby reducing their capability of differing from their counterparts in unrelated pseudogenes. This further diminishes the distinctiveness of suspected orthologous pairings.
Now consider flanking sequences. The earlier discussed fact that there is always some uncertainty in aligning of sequences18 implies that there must always be an element of doubt if ostensibly orthologous retropseudogenes are really located in exactly the same position in two or more genomes. In fact, it is acknowledged that the exact positions of many retroposed elements are uncertain or erroneous.89 Although primers can recognize presumably orthologous retropseudogene sequences whose flanking regions differ by as much as 25–30%,90 there are no absolute rules for the minimum degree of similarity required to justify such orthologous pairings.89 There are even published instances91 of orthologous pairings of LINE elements being accepted by several teams of investigators and then, upon reinvestigation, relocated hundreds of bases apart. Orthologous Alus, with dissimilarities in flanking sequences approaching 30%, are not limited to distantly-related primates, but are known to occur even in human-chimp comparisons, with the flanking repeats additionally differing in both base composition and overall length.87 In severe cases, the flanking regions of prospective orthologs are so dissimilar to each other that the orthologous pairing itself is doubtful.58
An unavoidable fudge factor is created by matching inexact sequences. There are even instances where the nucleotide differences in the presumed-orthologous flanking sequences actually form phylogenetically discordant groupings:
‘Thus, there is a C and an A shared by the gorilla and orangutan; a G shared by the baboon and rhesus; a C shared by the gorilla and pygmy chimpanzee; and a T shared by the orangutan and baboon. These examples of shared characters are discordant. The orangutan cannot have a recent common ancestry with the gorilla and with the baboon. The shared nucleotides can be interpreted as having arisen independently in two lineages. This raises the question of how many of such “shared nucleotides”, that have been used to support common ancestry, have actually arisen independently in two lineages(emphasis added)?’71
The flanking sequences which surround paralogous and orthologous retropseudogenes, already imprecisely similar to each other, are evidently not free to differ from each other in an unconstrained manner. An examination of three paralogous AS pseudogenes, each of which is compared to its orthologous pseudogene in different primates, indicates that flanking sequences vary from each other in a very nonrandom pattern of nucleotide substitutions that recur in parallel.92 This raises further doubts about the diagnostic uniqueness, in terms of nucleotide sequence, of each flanking sequence in the genome, as well as the belief that each such sequence is so unique that it (and its contained retropseudogene) can only be explained by shared evolutionary ancestry.
IV. Do orthologous pseudogenes have coincidental alterations?
To begin with, most pseudogenes contain multiple, nonunique alterations relative to their coding paralogs, making it often difficult to declare which one ostensibly inactivated the original gene.93 Moreover, orthologous primate pseudogenes can have different ‘inactivating mutations’.63 The fact that some orthologous human-chimp pseudogenes contain the same stop codon*6 appears impressive until one realizes that this is often not the case. For instance, a gorilla-specific CYP21 pseudogene has a stop codon while its indisputably-functional chimp ortholog does not and its human pseudogene ortholog does—but at a different location in the sequence.94 The CD8B1 gene provides another example of a gorilla-only stop codon.95 Elsewhere, a human OR pseudogene has a stop codon while its orthologous chimp pseudogene does not.96 And, when coincidental stop codons do occur, this is hardly compelling evidence for ‘shared mistakes’ in view of evidence for parallel nucleotide substitutions and parallel deletions (discussed later). The latter is relevant to frameshift-generated stop codons. Finally, we would expect coincidental stop codons because there are only three possibilities, and even these do not occur at subequal frequencies in pseudogenes.97
Nucleotide substitutions in pseudogenes, far from qualifying as ‘shared mistakes within the shared mistakes (pseudogenes)’, often contradict evolutionary schemes. The alpha-1,3-GT pseudogene, for instance, includes a nucleotide substitution at position 726 which is uniquely shared by cows, squirrel monkeys and gorillas.98 In the alpha-1,2-fucosyltransferase pseudogene,99 at position 258, the human and orang uniquely share a C, while chimp and gorilla uniquely share T. The rat and chimp uniquely share C at position 55 in the GLO pseudogene.100 Many nucleotide substitutions in the long Eta-globin pseudogene are either apomorphic or phylogenetically discordant.101 Orthologous Alu units of even closely related primates (e.g. humans and chimps) frequently exhibit considerable variance in nucleotide positions.87
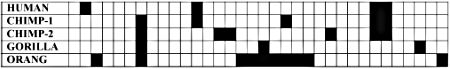
Figure 4.Schematic portrayal of non-corresponding (black) data observed when
nucleotide positions (columns) of orthologous pseudogenes (rows) are aligned. Indels
cause the misalignments.
Indels don’t fare much better, evolutionarily speaking. One can examine the 25,689 bases of the primate Beta-globin cluster (of which nearly half is the Eta-globin pseudogene) and quickly see that the vast majority of indels in the entire sequence are apomorphies. Furthermore, there are so many indels in the whole nearly-26 kb sequence [tabulated elsewhere22] that large ‘holes’ (Fig. 4) exist in the claimed sequence alignment of primates’ DNA. Still other indels are phylogenetically discordant. Although these include individual repetitive nucleotides, this fact must be put in perspective: some form of repetition is prevalent throughout even coding sequences.102
Elsewhere, a CYP chimp pseudogene has an 8 bp deletion not shared with its orang-utan, gorilla or human orthologous pseudogenes.94 A TPI chimp pseudogene has a long insertion* not found in its human orthologous pseudogene,103 while a DRB6 chimp pseudogene contains two insertions not shared with its human orthologous pseudogene.104 Not to be outdone, the gorilla ADPRTP1 pseudogene has a 30 bp duplicated region absent from its human orthologous pseudogene.105 In another instance, we observe a unique 6-base deletion/substitution sequence in the SHMT pseudogene undergoing a phylogenetic somersault: it is absent in the (ancestral) New World monkeys, present in the (more derived) Old World monkeys, and then is absent once again in the (most highly derived) apes and humans.106
Whether or not they occur only in pseudogenes, numerous molecular ‘shared events’ (mistakes or not), once considered virtually foolproof ‘perfect markers’ of evolutionary relatedness, have fallen victim to contrary evidence:
‘Nonetheless, almost every new molecular approach to phylogenetic inference has been ballyhooed as capable of “revolutionizing” the field … . Similar claims have been made for other kinds of data in the past. For instance, DNA-DNA hybridization data were once purported to be immune from convergence, but many sources of convergence have been discovered for this technique. Structural rearrangements of genomes were thought to be such complex events that convergence was highly unlikely, but now several examples of convergence in genome rearrangements have been discovered. Even simple insertions and deletions within coding regions have been considered to be unlikely to be homoplastic, but numerous examples of convergence and parallelism of these events are now known. Although individual nucleotides and amino acids are widely acknowledged to exhibit homoplasy, some authors have suggested that widespread simultaneous convergence in many nucleotides is virtually impossible. Nonetheless, examples of such convergence have been demonstrated in experimental evolution studies.’58
Of course, evolutionists still have faith (sic) in most if not all of these molecular markers. But they can hardly maintain any longer that common evolutionary descent is required to explain such things as ‘shared mistakes’.
V. Pseudogene-based phylogenies in the light of other evidence
| Autosomal pseudogene sequence: | Phylogeny: |
|---|---|
| Eta-globin | (Human–Chimp)Gorilla |
| UO | (Chimp–Gorilla)Human |
| Alpha-1,2 FT | (Chimp–Gorilla)Human |
| Alpha-1,3 GT | (Human–Chimp)Gorilla |
|
Table 2. The ambiguous sharing of “shared mistakes”. From Satta et al. (2000)
|
|
It has been asserted6 that evolutionary trees constructed on the basis of DNA similarities ‘agree remarkably well with the evolutionary trees derived earlier from anatomic similarities’. This statement is egregiously untrue. If anything, primate phylogenies are in a mess as a result of major contradictions between molecular and morphological data.57,107,108 Consider some recent craniodental data, which is very robust, statistically speaking. In a virtual mockery of pseudogene-based phylogenies (Fig. 2, Table 2), humans branch off first, followed by chimps, and finally a gorilla-orang clade*.108 (Gibbon was not considered in this study.)
Pseudogene-derived phylogenies are not even consistent with each other (Table 2). A common rationalization6 would have us believe that any difficulties in resolving the human-chimp-gorilla trichotomy have no impact on the validity of evolutionary theory itself. But consider the original prediction:
‘High expectations were placed on molecular methods, when these were first introduced, as to their power to resolve the trichotomy problem.’107
It is transparent special pleading to exalt molecular methods when they are predicted to support evolutionary notions, and then turn around and say that they are no threat to evolutionary theory when they fail! And, regardless of any post hoc rationalization invoked by the evolutionist to try to discredit it, the prima facie evidence (Table 2) refutes the claim that pseudogenes qualify as unambiguous shared mistakes among primates.
Of course, such inconsistencies are not limited to the H-C-G trichotomy. Barriel109 recently compared the previously-discussed Beta-globin data101 with 75 morphological elements, from another study, in order to construct a general primate phylogeny. The two data sets were found to conflict with each other, and so were ‘reconciled’ by being pooled together. The morphological data alone had placed the orang-utan as the sister group of the Homo/Pan/Gorilla clade (as in Fig. 2), but the pooled data displaced orang with the gibbon. In another study,110 Alu sequences were cited in support of the tarsier as the sister group of the anthropoid apes (and man), but this was acknowledged to contradict other phylogenies which place tarsiers elsewhere in the primate evolutionary tree. Overall, primate phylogenies constructed on the basis of retropseudogenes are not even confirmed by those based on other retroposons, the latter of which exhibit considerable phylogenetic conflicts among just themselves.111
Phylogenies based on ‘shared mistakes’ are not, of course, limited to primates, and the origin of whales has received much attention.6 Yet there are widely divergent phylogenetic inferences based on different lines of evidence.112 As usual, much evidence contradicting evolutionary relatedness is disregarded by the standard attribution to convergence. Apropos to the unconventional hippo-cetacean clade controversy, we are now in the proverbial situation of an irresistible force (pro: SINEs) encountering an immovable object (con: very strong skeletal evidence113). While some evolutionists insist that a favoured line of evidence trumps any dissenting evidence, other evolutionists warn against making such an assumption.114
All of the myriad problems with ‘convergent’ evolution, both molecular and morphological, are much too pervasive to be wished away as unimportant. If organic evolution is science, in the Popperian sense, and therefore subject to potential falsification, evolutionists must eventually acknowledge the fact that the overall profusion of divergent and contradictory phylogenies, pertaining to all forms of life, falsify macroevolution itself.
VI. Shared ‘mistakes’ without plagiarism or common ancestry
How written ‘plagiarized’ errors can arise without plagiarism
Phylogenetically-shared pseudogenes, as ‘shared mistakes’, have been compared to plagiarized written errors.6 A defendant was convicted of plagiarism by a court which recognized that, whereas similarity in books’ contents is to be expected from independently-acting authors writing about the identical topic, the same cannot be said about exact written errors. But this, of course, assumes the essential random nature of such errors, with concomitant extreme improbability of independent duplication. The court in question would have seen things differently had the ‘duplicated’ errors actually been only partly coincident from one book to another, especially if it was discovered that similar writing errors could arise independently after all.115 I will show that both considerations are very much applicable to pseudogenes.
Factors in the parallel deployment of pseudogenes
Figure 5 illustrates a retropseudogene insertion in its genomic context. In contrast to the assertion that processed pseudogenes are inserted at random locations into DNA,6 Miyamoto116 concludes that the tacit belief in the randomness of SINE insertion into the genome is ‘the least convincing assumption’ related to their role as phylogenetic markers. He cites evidences which show that specific target-site selection by retroelements is common. Let us develop this further, examining progressively finer levels of nonrandomness.
To begin with, lengthy Alu-barren intervals of host DNA are much more common than can be accounted for by a model which assumes constant probability of Alu insertion.117 It is hardly surprising that the density of Alu repeats, per kb of host DNA, varies widely according to location in the genome.118 Furthermore, Alu units often occur in clusters,119 even to the point of aggregating at almost the same orthologous position in different animals.120They are often found inserted, at the same spot, into each other.121,122 Evidence that the same site in the same primate is invaded repeatedly by Alus recognizably indicates that these are hotspots for Alu insertion,122 and the same holds for L1 insertions.123
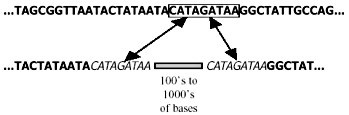
Figure 5.Two orthologous loci are illustrated: One (top) lacks a
retropseudogene, and the second (bottom) contains it (gray). Direct repeats are shown in
italics. These, and the flanking sequence, are shown identical for purposes of clarity.
Such is hardly ever the case.
The vast majority of Alus are located in the richest 40% (in terms of G+C) host DNA,124 and a disproportionate share of these insertions occur into 40–46% G+C host DNA.125 Both the tail and target regions are strongly enriched in A.126 There exists an astonishing positive correlation between (G+C) and CG-dimer* levels in Alus, or CG-dimer islands, and the (G+C) levels in the host DNA.127
The polynucleotide sequences located upstream some 10–20 sites from inserted Alu repeats and other retropseudogenes, are strongly biased towards certain hexamers*,128 and the same holds for L1 elements.129
Out of the 1024 (45) possible patterns of pentanucleotides* observed upstream from Alu repeats, only three of these are by far the most frequent.130 These, and successive, observations are recognized as evidence suggesting,131 and even indicating,132 site-specific insertions for retropseudogenes.
There exists a higher level of nonrandomness, one that is largely independent of, and therefore superimposed upon, the departures from randomness discussed thus far. Alu units are found concentrated in mitotic hotspots, early-replicating chromosomal bands, and other genomic locations.133 Moreover, the insertion of both LINEs and SINEs are believed to be strongly governed by the timing of chromosomal events.134 Locally, SINEs are believed to insert into existing breaks in the host DNA.135 Finally, experimental evidence136,137 demonstrates that there are very specific cleavage hotspots, for retropseudogene insertion, in bent or coiled DNA. All of these observations indicate that the widespread independent acquisition of interspersed elements (including retropseudogenes) is a workable proposition.
Can retropseudogenes be directly acquired by one individual organism from another? Some6 try to belittle the fact of horizontally-transmitted* genetic information as much as possible. But the list of known or strongly-suspected instances27 is now too large to be swept under the rug. Newer examples include the surprising discovery of SINE elements shared by distantly-related salmonid species,138 as well as between such evolutionarily-distant creatures as rodents and squids.15 There are also horizontally-shared LINE elements between vertebrate classes.139
Independently-originating variations within pseudogenes
It is not difficult to envision parallel occurrences of ‘shared mistakes’ because, as we have seen, coincidences between orthologous pseudogenes of different primates are, as a whole, very inexact. Also, as shown below, the similarities between indisputably unrelated pseudogenes is astonishing, and this indicates that only a limited number of degrees of freedom exist by which any given pseudogene can potentially differ from its paralogous gene, paralogous pseudogene(s), and/or orthologous pseudogene(s).
Consider some additional constraints: the DNA ‘alphabet’ consists of only 4 letters (bases), and the abundances of each nucleotide usually differ significantly from 25%,140 regardless of the etiology of the DNA sequence. Most pseudogenes, in comparison with their coding paralogs, are enriched in the following order: A>T>G>C.51 The same holds for Eta-globin pseudogene orthologs that are ‘progressively older’ insofar as they are shared by progressively more kinds of primates.141 Likewise, the inferred ‘mutational decay’ of AS pseudogenes shows a striking parallel pattern of nucleotide substitutions in different paralogous AS pseudogenes.92
Overall, transitional* nucleotide substitutions occur nearly twice as often as predicted by chance in pseudogenes.142 And, if there is a single base which differs from a consensus of 4 other orthologs, this nonconforming base is very likely to be a transition instead of a transversion*.143 Nor are the bases serially independent. For instance, if its right-side neighbour is G, the nucleotide C is particularly prone to vary, from pseudogene to pseudogene, as a transition.97 Nucleotide triplets also occur at strongly nonrandom frequencies.51
As with the example of lightning proved to strike twice, once it is shown that pseudogene alterations can happen independently but coincidentally, ‘shared mistakes’ no longer compel shared evolutionary ancestry. Evolutionists try to get around this by now arguing that genuine synapomorphies* invariably outnumber convergent ones. In most instances, this is a theory-driven assumption, because:
‘One can never tell whether two taxa share a nucleotide state by descent (homology) or chance (analogy).’71
More important, the common supposition that convergent molecular events occur too sporadically or disjointedly to account for the parallel deployment of ‘shared events’ (mistakes or not), in different organisms, is decisively contradicted by recent experimental evidence. Independent nucleotide substitutions144 and indels145,146 can occur in a sufficiently concerted manner to completely obscure accepted ancestor-descendant relationships.
The following is a rigorous example of evolutionists attempting to screen out the effects of convergence. This study101 involved an examination of the 17.2 kb sequence of the Eta-globin pseudogene that is shared by humans, chimps and gorillas. Among nucleotide substitutions, 12 parallel transitions and 7 transversions unique to human and chimps were found, compared to only 3 total substitutions exclusively shared by humans and distantly-related monkeys. Assuming a random distribution of substitutions, statistical analysis indicated that, at most, 7 of the 12, and 1 of the 7, of the said human-chimp synapomorphies could have arisen fortuitously. But such results do not compel an evolutionary origin because:
‘Naturally, these apparent synapomorphies could still have arisen separately under nonrandom conditions (e.g. if there were selective pressure in two species to preserve the same change, or a propensity of a nucleotide at a particular position to mutation in a particular direction). The simplest explanation, however, is that these changes are actual synapomorphies.’20
Now evolution of humans and chimps from a common ancestor has never been observed; nonrandom base substitutions and conserved orthologous base positions have manifested themselves countless times (and examples of both are reported in this work). So which explanation is simpler? Furthermore, it would take only a very weak common biasing effect (that is, a tiny deviation from randomness), imposed over such a long sequence (17.2 kb) to, at minimum, make up the difference between 7 and 12, and between 1 and 7.
Consider some constraints on pseudogene variance imposed by indels. From pooled data comprising 78 pseudogenes, it is evident that deletions are much more common than insertions. The size distribution of indels is strongly skewed, with over 50% of them only one base in length, and relatively few longer than five bases.8,92 The DNA content deleted from pseudogenes is itself nonrandom, consisting preferentially of repeated elements within short simple tandem arrays.147
Finally, with so many divergent and contradictory phylogenies in existence, at least one of them is bound to fortuitously coincide with the broad outlines of pseudogene deployment, and alteration, among primates. Consider also the following:
‘… the circularity of using inferred phylogenies to infer properties of molecular evolution that themselves influenced the reconstruction.’144
Alu units and their constrained differences
The repeated independent insertion of seemingly orthologous SINE units is facilitated by the (previously noted) fact that each SINE unit can potentially differ by only a very limited degree from another such unit. Were each Alu unit very different from another such unit, the chance of coincidental similarity in different primates, without common evolutionary descent, would be extremely small. Instead, Alus display an average global similarity of 70% to each other,148 and this rises to 81–98% within each Alu family’s respective consensus sequence.149
A ‘census’ of up-to 290 base positions150 shows that insertions within Alus are very nonrandom in terms of both the insertion’s position and length. As for nucleotide substitutions, hardly any of the 290 positions display less than a 70% preference for a particular base, with most of the remaining ≤30% dominated by one ‘second choice’. In fact, 195 positions are called CONSBI (conserved before insertion) because fewer than 14% of all Alus deviate from the preferred nucleotide at these positions.151 About half of the remaining sites (23 pairs, 46 total) consist of CG doublet hotspots which are prone to mutate frequently and (phylogenetically) unpredictably from one Alu element to another.83 For this reason, many investigators disregard these in phylogenetic analyses.
Such exclusion of nucleotides, however, only raises questions about both the paralogous and orthologous (phylogenetic) significance of the remaining ones. How do we know that the other so-called informative nucleotide substitutions are not also hotspots (albeit less extreme ones)? Nucleotide substitutions would then occur independently in primates in an apparently hierarchical manner, thus creating both the ‘Alu families’ and Alu-based phylogenies, but without making the hotspot locations as obvious. The earlier-discussed evidences for concerted parallel genomic alterations make the foregoing consideration all the more plausible. Moreover, there is evidence152 that nucleotide substitutions in the L1 during replication are nonrandom.
VII. Testing evolutionary claims
The factors governing pseudogene deployment and alteration, from primate to primate, are highly nonrandom. Consequently, assertions about the impossibility of independent shared ‘mistakes’6 are incorrect (Fig. 6). The only way that this conclusion could be contradicted would be through the performance of very detailed statistical tests which would examine all of the relevant factors.
A valid statistical test of retrospseudogenes must, at a minimum, take into account the following:
Figure 6.Schematic portrayal of the parallel acquisition of (inexact)
‘shared mistakes’, rendering a common evolutionary ancestry
unnecessary.
The fundamental overall nonrandomness (i.e. 50% random similarity in bases51) of the DNA molecule itself.
The ubiquitous presence of indels and resulting subjectivity in the alignment of units.
The liberties created by the after-the-fact invocation of missing loci.
The several different levels of nonrandomness pertaining to the insertion points themselves in the genome.
The large number of ‘trials’ (for independent ‘orthologous’ insertions) created by the vast number of known SINE units.
The fudge factor created by tolerating varying and often considerable amounts of sequence differences in the flanking sequences (and flanking repeats) when accepting them as orthologous.
The limited degree by which one SINE unit can differ from another,
The nonrandomness of nucleotide substitutions, indels, etc., in the retropseudogene unit itself.
Considerations 1–3, and 7–8, must likewise be tested in a manner that is relevant to classical pseudogenes.
Until such tests are performed, and rigorously substantiate the premise that classical pseudogenes cannot possibly originate from the independent disabling of orthologous genes in different organisms, and that retropseudogenes cannot be inserted independently in the same corresponding locations in different primates, evolutionistic arguments about shared ‘mistakes’6 should not be given credence.
Not enough is yet known about eukaryotic genomes to construct a comprehensive creationist model of pseudogenes. Nevertheless, the belief that ‘pseudogenes are unequivocal support for evolution’6 is invalid. New evidence is constantly being published that weakens or invalidates one or other long-held evolutionistic beliefs about pseudogenes. Now, more than ever, it is an exciting time to be a creationist scientist.
Acknowledgements
I thank the following individuals for advice and review: Dr Paul Nelson, Dr Jerry Bergman, Dr David DeWitt, and Dr Michael Brown.
Glossary
Alu—A category of well-known SINEs. Return to text.
Antisense RNA—RNA which copies the DNA from the reverse direction. Return to text.
Apomorphy—A trait which is unique to the organism in question. It is not shared with either ‘less derived’ or ‘more derived’ organisms. Return to text.
Base—Denoting the 4 biochemicals (A—Adenine, G—Guanine, C—Cytosine, T—Thymine (U—Uracil in RNA)) that are part of a nucleotide. The information to code for proteins can be stored in sequences of bases. Return to text.
bp— Abbreviation for base-pair. Return to text.
Clade—A branching-off point of an organism or closely-related set of organisms relative to presumably-ancestral organisms. Return to text.
Convergence—The acquisition, by organisms, of shared traits independently (without having inherited them from a shared evolutionary ancestor). Return to text.
Deletion—The removal of a segment of the DNA sequence followed by reconnection of the free ends of the molecular ‘chain’. Compare Insertion. Return to text.
Dimer—An association of two Bases. Return to text.
Direct Repeats—That part of the Flanking Sequence which is duplicated prior to the insertion of the retropseudogene. See Fig. 5. The direct repeats are illustrated in italics. Return to text.
Eukaryotes—Organisms which have an organized cell nucleus. All living things, except bacteria and archarbacteria, are eukaryotes. Return to text.
Flanking Sequence—That part of the DNA ‘chain’ which immediately precedes, and immediately comes after, a retropseudogene. See Fig. 5. Return to text.
Gene Conversion—The process whereby one gene is used as a template to ‘overprint’ another. The latter thereby is forced to resemble the former. Return to text.
Hexamer—A string of six Bases. Return to text.
Homoplasy—Convergence and Parallelism. Return to text.
Homopolymer—A chain of identical bases: AAAAA … , CCCCC … , GGGGG … , or TTTTT … . Return to text.
Horizontal Transmission—The direct transmission of genetic information from one living individual to another. Return to text.
Indel—Acronym for Insertion or Deletion. See Fig. 4. Return to text.
Insertion—The addition of a new segment of the DNA sequence followed by reconnection of the free ends of the molecular ‘chain’. Compare Deletion. Return to text.
Intergenic—Occuring on the DNA molecule between genes. Return to text.
Interspersed Repeats—A group of genomic elements which occur in great profusion. Notable interspersed repeats are LINEs and SINEs. Return to text.
kb—Abbreviation for kilobase; 1000 Bases. Return to text.
L1—A group of well-known LINEs. Return to text.
LINE—Long interspersed nuclear element. A group of retropseudogenes that occur in the hundreds of thousands in the human genome, and which are typically about 7,000 bases long. Return to text.
Locus (Loci)—A specific position on a chromosome. Return to text.
Nested Hierarchy—A series of progressively narrowly-defined subsets which reflect presumably-increasing evolutionary derivation. For example, a member of the vertebrates gave rise to mammals, a member of the mammals gave rise to primates, and a member of the primates gave rise to humans. See Fig. 2 for an ‘advanced’-primate nested hierarchy. Return to text.
Nucleotide—A compound of a sugar, phosphate and base—DNA and RNA comprise of nucleotides. Return to text.
Ortholog—Gene and/or pseudogene which is a counterpart to a similar gene and/or pseudogene in another primate. An ortholog is presumed to be a copy of an ancestral gene sequence. Refer to Fig. 1. Compare Paralog. Return to text.
Parallelism—The acquisition, by organisms, of shared traits independently (without having inherited them from a shared evolutionary ancestor). See Fig. 6. Return to text.
Paralog—Copy of the same gene, pseudogene, etc. within the same organism. See Fig. 1. Compare Ortholog. Return to text.
Pentanucleotide—A chain of five Nucleotides. Return to text.
Phylogen(-ic, -y)—Related to the construction of an evolutionary ‘tree’. Return to text.
Poly-A—Consisting of many adenine bases in succession: AAAAAAAA … . Return to text.
Poly-A tail—A sequence of adenine bases at the end of an RNA molecule or a pseudogene. Return to text.
Purine—The Bases adenine (A) and guanine (G). Return to text.
Pyrimidine—The Bases cytosine (C) and thymine (T). Return to text.
Retro- (-element, -poson, -pseudogene)—A (given structure) created by the reverse transcription (in effect, ‘backfiring’) of RNA back into the host DNA. Return to text.
SINE—Short interspersed nuclear element. A group of retropseudogenes that occur in the hundreds of thousands in the human genome, and each of which is typically about 300 bases long. Return to text.
Stop codon—A triplet of Nucleotides which puts a stop to protein synthesis. Return to text.
Synapomorphy—A trait which is shared by two or more organisms, and which supposedly is the result of a recent common evolutionary ancestor. Return to text.
Tail—see Poly-A tail. Return to text.
Transition—In the DNA molecule, the replacement of one Purine by another Purine, or the replacement of one Pyrimidine by another Pyrimidine. Compare Transversion. Return to text.
Transversion—In the DNA molecule, the replacement of a Purine by a Pyrimidine, or vice-versa. Compare Transition. Return to text.
References and notes
- Zuckerkandl, E. and Hennig, W., Tracking heterochromatin, Chromosoma 104:75, 1995.
- Zuckerkandl, E. et al., Maintenance of function without selection, J. Molecular Evolution 29:504, 1989.
- A pseudogene can be likened to a wheel-less automobile. But, as we shall see, immobility need not imply nonfunction. Throughout this work, I use ‘evolspeak’ for purposes of clarity. However, I would like to see nonprejudicial language emerge (e.g. genoid instead of pseudogene, nucleotide variance instead of nucleotide substitution or inactivating mutation, etc.).
- Moreira, M.A.M. and Seuanez, H.N., Mitochrondrial pseudogenes and phyletic relationships of Cebuella and Callithrix (Platyrrhini, Primates), Primates 40(2):353–364, 1999.
- Gibson, C.J., Pseudogenes and origins, Origins (Loma Linda) 2(2):91–108, 1994. This article is from a scientific creationist viewpoint.
- Max, E.E., Plagiarized errors and molecular genetics, <www.ics.uci.edu/pub/bvickers/origins/molecular-genetics.txt>. (Last update: 12 July 1999). Also <www.talkorigins.org/faqs/molgen> (Last update: 6 June 2000). For years, Max has argued that pseudogenes, as ‘shared mistakes’ between primate genomes, constitute unequivocal evidence against special creation and for organic evolution.
- Esnault, C. et al., Human LINE retrotransposons generate processed pseudogenes, Nature Genetics 24:363, 2000. It is currently supposed that master gene(s), rather than retroviruses, reverse-transcribe themselves into the DNA, thus generating SINEs and LINEs as pseudogenes.
- Gu, X. and Li, W-H., The size distribution of insertions and deletions in human and rodent pseudogenes suggests a logarithmic gap penalty for sequence alignment, J. Molecular Evolution 40:465–469, 1995.
- Thorne, J.L. and Kishino, H., Freeing phylogenies from artifacts of alignment, Molecular Biology and Evolution 9(6):1150–1151, 1992.
- Li, W-H. et al., Pseudogenes as a paradigm of neutral evolution, Nature 292:237, 1981. The authors match a mouse gene with its inferred pseudogene paralog, disregarding a 30-nucleotide non-corresponding segment, which is blamed on an insertion.
- Nishikimi et al., Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, J. Biological Chemistry 269(18):13686, 1994.
- Kondrashov, A.S. and Koonin, E.V., Molecular evolution in the genomic era, Cell 101(2):128–129, 2000.
- Cavalier-Smith, T. and Beaton, M.J., The skeletal function of nongenic nuclear DNA, Genetica 106:3–13, 1999. Of course, no-one is invoking Lamarckianism, wherein an organism could somehow communicate with its genome and, in this instance, expel useless DNA on command.
- Farlow, B., Stuff or nonsense? New Scientist 166(2232):38–41, 2000.
- Ohshima, K. et al., Several short interspersed repetitive elements (SINEs) in distant species may have originated from a common ancestral retrovirus, Proc. Nat. Acad. Sci. USA 90:6260–6264, 1993.
- Mager, D.L., Endogenous retroviruses provide the primary polyadenylation signal for two new human genes (HHLA2 and HHLA3), Genomics 59:255, 1999. On the other hand, the statement that many harmful pseudogenes have existed at one time or another, but that all but the most recently-originated harmful ones have been removed from populations by natural selection, is little more than an evolutionary and long-age supposition.
- Mighell, A.J., Vertebrate pseudogenes, FEBS Letters 468:113, 2000.
- Lomax., M.I. et al., Rapid evolution of the human gene for cytochrome C oxidase subunit IV, Proc. Nat. Acad. Sci. USA 89:5269, 1992.
- Minghetti, P.P. and Dugaiczyk, A., The emergence of new DNA repeats and the divergence of primates, Proc. Nat. Acad. Sci. USA 90:1875, 1993.
- Fitch, D.H.A. et al., The spider monkey eta-globin (pseudo) gene and surrounding sequences, Genomics 3:237–250, 1988.
- Ophir, R. and Graur, D., Patterns and rates of indel evolution in processed pseudogenes from humans and murids, Gene 205:199–201, 1997.
- Saitou, N. and Ueda, S., Evolutionary rates of insertion and deletion in noncoding nucleotide sequences of primates, Molecular Biology and Evolution 11(3):504–511, 1994.
- Freytag, S.O. et al., Molecular structures of human argininosuccinate synthetase pseudogenes, J. Biological Chemistry 259:3165, 1984. This disparity is rationalized away by the ad hoc suggestion that the parent gene has mutated after the pseudogenes had diverged from it. Supposedly, the 19 unique pseudogene nucleotide substitutions reflect the state of the parent gene prior to the divergence.
- One of the authors of this paper will be Dr Paul Nelson, Ph.D. in Philosophy with emphasis on biology, from the University of Chicago. Nelson is active in the intelligent design movement.
- McCarrey, J.R. and Riggs, A.D., Determinator-inhibitor pairs as a mechanism for threshold setting in development: a possible function for pseudogenes, Proc. Nat. Acad. Sci. USA 83:679–683, 1986. Pursuing the earlier analogy of the pseudogene to a wheel-less car, the latter actually has a nontransportation function (its motor/transmission turns a thresher). The authors have informed me that no one has as yet tested their theory.
- Schmid, C.W. and Shen, C-K.J., The evolution of interspersed repetitive DNA sequences in mammals and other vertebrates; in: Molecular Evolutionary Genetics, Plenum Press, New York, pp. 332–337, 348–351, 1985.
- Walkup, L.K., Junk DNA, TJ 14(2):18–30, 2000.
- Hamdi, K.H., et al., Alu-mediated phylogenetic novelties in gene regulation and development, J. Molecular Biology 299(4):931–939, 2000.
- Li, W-H., Molecular Evolution, Sinauer, Massachusetts, USA, p. 347, 1997.
- Woodmorappe, J., The Mythology of Modern Dating Methods, Institute for Creation Research, El Cajon, California, p. 3, 1999.
- Here’s a facetious example: The evolutionist first says that pseudogenes have no function. When a function is discovered, he zeroes in on the technicality of the pseudogene being green and says: ‘Function may be applicable to green pseudogenes, but to no others!’ When, however, function emerges for striped pseudogenes, the evolutionist changes his tune and says: ‘Well, green pseudogenes and striped ones have functions, but this doesn’t really mean anything.’ Time goes on, and a function turns up for polka-dotted pseudogenes. So we now hear: ‘Pseudogenes are functionless, with the minor exception of green, striped and polka-dotted ones.’ And so on ad infinitum.
- Max, E., Reply to Wieland, <www.talkorigins.org/faqs/molgen/wieland.html>.
- Wieland, C., ‘Junk-making’ viruses neutralize an evolutionary argument, TJ 10(3):296–297, 1996.
- Dimitri, P. and Junakovic, N., Revising the selfish DNA hypothesis, Trends in Genetics 15(4):123–124, 1999.
- An example of this would be an inspection of a warehouse wherein 1,000 boxes of fruit are stored. All 1,000 are declared by the owner to be vermin-free. An inspector opens 5 boxes, and finds vermin. Following the ATM fallacy, as practiced by Max6 in relation to pseudogene function, the owner could plead: ‘You have not shown that the overwhelming majority of boxes (the 995) contain vermin!’ Obviously, this will not do. A reasonable suspicion now surrounds the remaining 995 boxes and, short of examining them all, the burden of proof now shifts to the owner to defend their wholesomeness. In like manner, the discoveries of functional pseudogenes create a reasonable suspicion about the (allegedly) nonfunctional majority. Short of an examination of every single pseudogene (even this would likely be inconclusive), the burden of proof now shifts to those who continue to advocate overall pseudogene nonfunction.
- Popper, K.R., The Logic of Scientific Discovery, Basic Books, New York, pp. 87, 315, 1959. In his classic example, the assertion that all ravens are black is falsified by observing only one white raven. Now consider the popular (but erroneous) belief that lightning striking twice in one spot is infinitesimally improbable. How many instances of lightning striking at different localities will prove this? None—because, as Popper emphasizes, theories are not validated by any amount of congenial evidence. How many times must lightning strike twice to disprove this? Exactly one. And, after that, it becomes pointless to demand more examples, or to quibble about whether lightning could strike the same location twice or even 20 times. The ATM fallacy allows evolutionists, in essence, to recognize one tree after another and yet refuse to admit that they are in a forest. Essentially the same fallacy occurs when evolutionists assert6 that the discovery of functional pseudogenes does not threaten the supposition that most pseudogenes are useless. Really!
- Weiner, A.M., Do all SINEs lead to LINEs? Nature Genetics 24:333, 2000.
- Makalowski, W., SINEs as a genomic scrap yard; in: The Impact of Short Interspersed Elements (SINEs) on the Host Genome, R.G. Landes Co., Texas, p. 81, 1995.
- von Sternberg, R.M. et al., Genome canalization, Genetica 86:216, 1992.
- Howard, B.H. and Sakamoto, K., Alu interspersed repeats: selfish DNA or a functional gene family? New Biologist 2:759, 766, 1990.
- Fedoroff, N.V. Transposable elements, Annals of the New York Academy of Sciences 870:256, 1999.
- Tachida, H. and Iizuka, M., A population genetic study of the evolution of SINEs, Genetics 133:1023, 1993.
- Gabriel, A., Transposons in the human genome, in: Molecular Biology and Biotechnology, VCH Publishers, New York, NY, p. 928, 1995.
- Fanning, T.G. and Singer, M.F., LINE-1: a mammalian transposable element, Biochimica et Biophysica Acta 910:209, 1987.
- Petrov, D.A. and Hartl, D.L., Pseudogene evolution and natural selection for a compact genome, J. Heredity 91:222, 2000. These authors present the intriguing theory that pseudogenes have position effects on the expression of nearby genes.
- Panning, B. and Smiley, J.R., Activation of RNA polymerase III transcription of Human Alu elements by Herpes simplex virus, Virology 202:408, 1994.
- Britten, R.J., Mobile elements inserted in the distant past have taken on important functions, Gene 205:181, 1997. His (now too small) list of 21 known functional mobile insertions includes 7 Alu elements.
- Piedrafita, E.J. et al., An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element, J. Biological Chemistry 271:14419, 1996.
- Schmid, C.W. and Rubin, C.M., Alu: What’s the use? in: Makalowski, Ref. 38, p. 106.
- Consider this illustrative counter-example: A conviction rests solely on the results of one forensic technique. Recent evidence proves that it sometimes implicates innocent people. In the future, this technique will likely be ruled inadmissible in court. But the defendant is not grasping at some wished-for future development: he is citing the current state of affairs, which has already created a reasonable doubt about his guilt, and for which reason he should be acquitted. In like manner, more than a reasonable doubt already exists about generalized ‘nonfunctional pseudogene’ beliefs.
- Blake, R.D. et al., The influence of nearest neighbors on the rate and pattern of spontaneous point mutations, J. Molecular Evolution 34:190–196, 1992.
- Grewal, P.K. et al., Recent amplification of the human FRG1 gene during primate evolution, Gene 227:85, 1999. My Figure 1 illustrates only a few possibilities. Real primate genomes are vastly more complex.
- Bailey, A.D. et al., Molecular origin of the mosaic sequence arrangements of higher primate alpha-globin duplication units, Proc. Nat. Acad. Sci. USA 94:5179–5180, 1997.
- Collura, R.V. and Stewart, C-B., Insertions and duplications of mtDNA in the nuclear genomes of Old World monkeys and hominoids, Nature 378:487, 1995.
- Ueda, S. et al., A truncated immunoglobulin Epsilon-pseudogene is found in gorilla and man but not in chimpanzee, Proc. Nat. Acad. Sci. USA 82:3712–3713, 1985.
- Ueda, S. et al., Multiple recombinational events in primate immunoglobulin epsilon and alpha genes suggest closer relationship of humans to chimpanzees and gorillas, J. Molecular Evolution 27:77–83, 1988.
- Revolo, Molecular phylogeny of the hominoids, Molecular Biology and Evolution 14(3):248–265, 1997.
- Hillis, D.M., SINEs of the perfect character, Proc. Nat. Acad. Sci. USA 96:9979–9980, 1999. Whenever a significant fraction of loci are missing in a phylogenetic comparison of several organisms, there is no way to determine whether the members of the taxa in question ever contained the inserted element. The hierarchical sharing of certain inserted elements then becomes untestable. As a result, missing loci unavoidably introduce a fudge factor relative to any evaluation of ‘shared mistakes’.
- Sharon, D. et al., Primate evolution of an olfactory receptor cluster:
diversification by gene conversion and recent emergence of pseudogenes, Genomics
61:27–32, 1999. Even though this situation is decided by one-base deletions,
it must be remembered that (as discussed elsewhere) the vast majority of deletions in
pseudogenes are only one base long. In a broader context, the cluster of genes and
pseudogenes which comprise the Olfactory Receptor (OR) cluster do not show
straightforward hierarchical deployment. Various conversion events (including fused genes
and duplicated genes) are each apomorphic to humans and chimps. Two gene conversion
events occur in the same location in monkey chromosomes, and this is attributed to a hot
spot in the genome.
If one examines the overall percentage of the primate OR gene repertoire that is occupied by pseudogenes, one does observe a crude increase in percentage relative to increasingly-derived infra-orders of primates. But this crude progression breaks down as soon as one includes the prosimians. These least-derived primates have a percent pseudogene content which overlaps that of even the highly-derived hominoids. Rouquier, S. et al., The olfactory receptor gene repertoire in primates and mouse, Proc. Nat. Acad. Sci. USA 97:2873, 2000.
- Qin, Z. et al., The interleukin-6 gene locus seems to be a preferred target site for retrotransposon integration, Immunogenetics 33:265, 1991.
- Martignetti, J.A. and Brosius, J., BC200 RNA, Proc. Nat. Acad. Sci. USA 90:11565–11566, 1993. The gene conversion event supposedly created the human-only pseudogene after the human-chimp split.
- Fitch, H.A. et al., Duplication of the gamma-globin gene mediated by L1 long interspersed repetitive elements in an early ancestor of simian primates, Proc. Nat. Acad. Sci. USA 88:7396–7398, 1991.
- Kriener, K. et al., Convergent evolution of major histocompatibility
molecules in humans and New World monkeys, Immunogenetics 51:169–178,
2000.
Elsewhere, an even more conspicuous absence of a common set of gene-inactivating mutations occurs in the delta-globin and psi-etaglobin pseudogenes of the Old World monkeys. Far from being ‘shared mistakes’, the various ostensibly gene-silencing frameshifts, deletions, and point mutations are each unique to rhesus, colubus, and the baboon: Vincent, K.A. and Wilson, A.C. Evolution and transcription of Old World Monkey globin genes. J. Molecular Biology 207:466, 478, 1989.
- Kim, J-H. et al., Unique sequence organization and erythroid cell-specific nuclear factor-binding of mammalian theta-1 globin promoters, Nucleic Acids Research 17:5687–5691, 1989. This state of affairs is credited to an assumed inactivation of the theta-1 globin genes early in primate evolution, followed by the evolutionary divergence of lower and higher primates, and finally the eventual secondary reactivation of the theta-1 globin genes in the more-derived higher primates but not in the less-derived lower primates.
- Vallinoto, M. et al., Mitochrondrial DNA-like sequence in the nuclear genome of Saguinus (Callitrichinae, Primates), Genetics and Molecular Biology 23:35–42, 2000.
- Maeda, N. et al., Molecular evolution of intergenic* DNA in higher primates, Molecular Biology and Evolution 5:2, 1988.
- Mindell, D.P., Multiple independent origins of mitochondrial gene order in birds, Proc. Nat. Acad. Sci. USA 95:10693–10697,1998.
- Woodmorappe, J., Noah’s Ark: A Feasibility Study, Institute for Creation Research, El Cajon, California, pp. 202–209, 1996.
- Kawaguchi, H., C4 genes for the chimpanzee, gorilla, and orangutan, Immunogenetics 35:16–23, 1992b.
- Bergstrom, T.F. et al., Recent origin of HLA-DRB1 alleles and implications for human evolution, Nature Genetics 18:237–242, 1998.
- Hamdi, H. et al., Origin and phylogenetic distribution of Alu DNA repeats, J. Molecular Biology 289:866–867, 1999.
- Boeke, J.D., LINEs and Alus—the polyA connection, Nature Genetics 16:7, 1997.
- Murata, S. et al., Details of retropositional genome dynamics that provide a rationale for a generic division, Genetics 142:922–923, 1996. In this study, no less than 70% of orthologous loci were found to be missing.
- Smit, A.F.A. et al., Ancestral, mammalian-wide subfamilies of LINE-1 repetitive sequences, J. Molecular Biology 246:406–407, 1995.
- Westhoff, C.M. and Wyllie, D.E., Investigation of the RH locus in gorillas and chimpanzees, J. Molecular Evolution 42:667–668, 1996.
- Leeflang, E.P., Phylogenetic isolation of a human Alu flounder [sic] gene, J. Molecular Evolution 37:563–565, 1993a.
- Kass, D.H., Gene conversion as a secondary mechanism of short interspersed element (SINE) evolution, Molecular and Cellular Biology 15:19–25, 1995.
- Leeflang, E.P. et al., Mobility of short interspersed repeats within the chimpanzee lineage, J. Molecular Evolution 37:566–568, 1993b.
- Cantrell, M.A. et al., An ancient retrovirus-like element contains hot spots for SINE insertion, Genetics (In Review), 2001.
- Slattery, J.P. et al., Patterns of diversity among SINE elements from three Y-chromosome genes in carnivores, Molecular Biology and Evolution 17(5):825–829, 2000. Not surprisingly, some evolutionists have challenged these findings.6 But others have accepted them.
- Burton, F.H. et al., L1 gene conversion or same-site transposition, Molecular Biology and Evolution 8(5):609–619, 1991. An alternative interpretation suggested by the authors is that the L1 insertions are synapomorphic* after all, but have undergone gene conversion subsequent to their emplacement. In that case, the absence of this insertion at the locus of an intermediately-derived rodent must be explained away somehow. The authors have considered precise excision of the L1 unit, or perhaps the presence of the insertion exclusively at a chromosome that did not last in the population of the intermediately-derived rodent.
- Leeflang, E.P. et al., Phylogenetic evidence for multiple Alu source genes, J. Molecular Evolution 35:12–15, 1992. At the time, the Ya5 Alu family was called PV (Precise Variant), or HS (Human-Specific), which it isn’t.
- Yang, A.S., The rate of CpG mutation in Alu repetitive elements within the p53 tumor suppressor gene in the primate germline, J. Molecular Biology 258:240–250, 1996.
- Boissinot, S. et al., L1 (LINE-1) retrotransposon evolution and amplification in recent human history, Molecular Biology and Evolution 17:924, 2000.
- Batzer, M.A. et al., Standardized nomenclature for Alu repeats, J. Molecular Evolution 42:4-5, 1996.
- Filipski, J. et al., Chromosome location-dependent compositional bias of point mutations in Alu repetitive sequences, J. Molecular Biology 206:564–565, 1989.
- Sawada, I. et al., Evolution of Alu family repeats since the divergence of human and chimpanzee, J. Molecular Evolution 22:318–319, 1985.
- Daniels, G.R. and Deininger, P.L., Integration site preferences of the Alu family and similar repetitive DNA sequences, Nucleic Acids Research 13:8941–8943, 1985.
- Jurka, J., Approaches to identification and analysis of interspersed repetitive DNA sequences; in: Adams, M.D. et al., Automatic DNA Sequencing and Analysis, Academic Press, London, New York, pp. 295–296, 1994.
- Shedrock, A.M. and Okada, N., SINE insertions, BioEssays 22:154, 2000.
- Smit, A.F.A., Structure and Evolution of Mammalian Interspersed Repeats, Ph.D. Dissertation, University of Southern California, pp. 130–135, 1996.
- Casane, D. et al., Mutation pattern variation among regions of the primate genome, J. Molecular Evolution 45:219–221, 1997.
- Li, W-H., Evolution of duplicate genes and pseudogenes; in: Evolution of Genes and Proteins, Sinauer Associates, Massachussets, pp. 30–32, 1983.
- Kawaguchi, H. et al., Evolutionary origin of mutations in the primate cytochrome P450c21 gene, American J. Human Genetics 50:776–777, 1992a.
- Delarbre, C. et al., Duplication of the CD8 Beta-chain gene as a marker of the man-gorilla-chimpanzee clade, Proc. Nat. Acad. Sci. USA 90:7050, 7052, 1993.
- Glusman, G. et al., Sequence, structure, and evolution of a complete human olfactory gene cluster, Genomics 63:230, 2000.
- Bulmer, M., Neighboring base effects on substitution rates in pseudogenes, Molecular Biology and Evolution 3(4):324–327, 1986. TGA is by far the most commonly occurring stop codon. Furthermore, out of 64 possible codons in large human genes, only 18 of these (of which 11 are common) are vulnerable to conversion into a stop codon by a single nucleotide substitution: Modiano, G., Nonrandom patterns of codon usage and of nucleotide substitutions in human alpha and beta-globin genes, Proc. Nat. Acad. Sci. USA 78:1122, 1981. Kimura, M. The Neutral Theory of Molecular Evolution, Cambridge University Press, pp. 183–185, 1983. When the cited large human genes are examined for actual abundances of all possible codons, and the eventual TGA stop codon-bias is taken into account, the limited possibilities for stop codon occurrence become all the more obvious. Only 2–4% of the individual codons in use are within one nucleotide substitution of becoming TGA, the majority-occurring stop codon in human pseudogenes. Of course, since duplicate gene copies presumably change into pseudogenes, the positions of the 2–4% progenitor codons are fixed in each copy. Finally, it is acknowledged that identical stop codons occurring at the same location in potentially-orthologous pseudogenes need not imply shared evolutionary ancestry. Such is the case with human and sheep P2 pseudogenes, for which coincidental stop codons are believed to be of independent origins: Medd, S.M. and Walker, J.E., Evolution of the expressed P2 pseudogene and the origin of the P1 and P2 genes, Biochemical Journal 293:73, 1993.
- Galili, U. and Swanson, K., Gene sequences suggest inactivation of Alpha-1,3-galactosyltransferase in catarrhines after the divergence of apes from monkeys, Proc. Nat. Acad. Sci. USA 88:7403, 1991.
- (No listed author), Pseudogene for alpha-(1,2)-fucosyltransferase, <sayer.lab.nig.ac.jp/~silver/apeData/pfut2/pfut2.html>, July 2000.
- Ohta, Y. and Nishikimi, M., Random nucleotide substitutions in primate nonfunctional gene for L-gulono-gamma-lactone oxidase, Biochimica et Biophysica Acta 1472:409, 1999.
- Bailey et al., Reexamination of the African hominoid trichotomy with additional sequences from the primate Beta-globin gene cluster, Molecular Phylogeny and Evolution 1:115–132, 1992. For example, at position 984, a (T) is uniquely shared by both the species of chimp and the species of distantly related galago. As for indels, a deletion spanning positions 5098–5101 is shared only by the phylogenetically-distant orang and spider monkey. This example does not include anomalous clustering of indels at homopolymeric sites, where independent generation of indels is a common occurrence.
- Gusev, V.D. et al., On the complexity measures of genetic sequences, Bioinformatics 15(2):994, 1999.
- Craig, L.C. et al., Characterization of the transcription unit and two processed pseudogenes of chimpanzee triosephosphate isomerase (TPI), Gene 99:225, 1991.
- Figueroa, F. et al., Primate DRB6 pseudogenes, Immunogenetics 34:335, 1991.
- Lyn, D. et al., Conservation of sequences between human and gorilla lineages, Gene 155:243, 1995.
- Devor, E. et al., Serine hydroxymethyltransferase pseudogene, SHMT-ps1, J. Experimental Zoology 282:153, 156, 1998. A more flagrant instance of a ‘shared mistake’ that cannot be the result of common ancestry is as follows: Two HLA genes and three HLA pseudogenes in the human genome share an identical deletion. Because a single ancestral gene is unlikely for this motley group of five, a veritable dance of ad hoc gene conversion events is invoked. We are asked to imagine that the two genes and three pseudogenes ‘passed on’ the identical deletion to each other, each time involving only the short segment of the DNA that surrounds the deletion, and that they accomplished this perhaps several times, Geraghty, D.E. et al., Examination of four HLA Class I pseudogenes, J. Immunology 149:1954–1955, 1992.
- Satta, Y. et al., DNA archives and our nearest relative, Molecular Phylogenetics and Evolution 14(2):259–275, 2000.
- Collard, M. and Wood, B., How reliable are human phylogenetic hypotheses? Proc. Nat. Acad. Sci. USA 97:5003–5006, 2000.
- Barriel, V., Pan paniscus and hominoid phylogeny, Folia Primatologica 68:50–56, 1997.
- Zietkiewicz, E. et al., Phylogenetic affinities of tarsier in the context of primate Alu repeats, Molecular Phylogenetics and Evolution 11(1):77, 1999.
- Johnson, W.E. and Coffin, J.M., Constructing primate phylogenies from ancient retrovirus sequences, Proc. Nat. Acad. Sci. USA 96:10254–10260, 1999. In a complete breakdown of any semblance to an evolutionary nested hierarchy, HERV-K(C4) is present in the (less derived) Old World monkeys, and in some apes, but is anomalously absent at the expected loci in the (highly-derived) gorillas and chimps. For this reason, a gene-conversion rationalization is invoked.
- O’Leary, M.A., Parsimony analysis of total evidence from extinct and extant taxa and the Cetacean-Artiodactyl question (Mammalia, Ungulata), Cladistics 15:315–330, 1999.
- Luckett, W.P. and Hong, H., Phylogenetic relationships between the orders Artiodactyla and Cetacea, J. Mammalian Evolution 5:133–143,154–160, 169, 1998.
- Gura, T., Bones, molecules … or both? Nature 406:230–233, 2000.
- Obvious examples include: striking adjacent keys (‘r’ vs. ‘t’), inclusion of commonly-misspelled words, and use of common grammatical errors. Less intuitively-obvious ones are: typists independently making the same nonadjacent-key typos owing to typewriter-key mechanics and/or the physiological mechanics of human fingers; inexperienced printers botching the same long words in the same way; fatigued writers slipping into very similar written errors owing to common perceptual and neuromental processes.
- Miyamoto, M.M., Perfect SINEs of evolutionary history? Current Biology 1999(9):R816, 1999.
- Moyzis, R.K. et al., The distribution of interspersed repetitive DNA sequences in the human genome, Genomics 4:276–281, 1989.
- Beck, S., Evolutionary dynamics of non-coding sequences within the Class II region of the human MHC, J. Molecular Biology 255:6, 1996.
- Makalowski, W. et al., Alu sequences in the coding regions of mRNA, Trends in Genetics 10(6):188, 1994.
- Lee, Y. et al., Complete genomic sequence and analysis of the prion protein gene region from three mammalian species, Genome Research 8:1025, 1033, 1998. These almost-coincident Alus, inserted at the same hotspot region of the genome, may be understood as ‘near misses’ in the independent orthologous insertions of Alus.
- Glusman, G. et al., Genome dynamics, polymorphisms, and mutations in a 350 kb human olfactory receptor gene cluster, Mathematical Modelling and Scientific Computing 9:30–44, 1998.
- Bailey, A.D. and Shen, C-K.J., Sequential insertion of Alu family repeats into specific genomic sites of higher primates, Proc. Nat. Acad. Sci. USA 90:7205-7208, 1993.
- DeBerardinis, R.J. and Kazazian, H.H., Full-Length L1 elements have arisen recently in the same 1 kb region of the gorilla and human genomes, J. Molecular Evolution 47:300, 1998.
- Smit, A., The origin of interspersed repeats in the human genome, Current Opinion in Genetics and Development 6:744, 1996.
- Smit, A., Interspersed repeats and other mementos of transposable elements in mammalian genomes, Current Opinion in Genetics and Development 9:658, 1999.
- Jurka, J. and Klonowski, P., Integration of retroposable elements in mammals: selection of target sites, J. Molecular Evolution 43:689, 1996.
- Jabbari, K. and Bernardi, G., CpG doublets, CpG islands and Alu repeats in long human DNA sequences from different isochore families, Gene 224:126–127.
- Jurka, J., Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons, Proc. Nat. Acad. Sci. USA 94:1875, 1997.
- Cost, G.J. and Boeke, J.D., Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure, Biochemistry 37:18089–18090, 1998.
- Toda, Y. et al., Characteristic sequence pattern in the 5- to 20-bp upstream region of primate Alu elements, J. Molecular Evolution 50:235, 2000.
- Toda, Y. et al., Sequence patterns observed in 5' flanking regions of primate Alu elements, Annals of the New York Acad. Sci. 870:372–373, 1999.
- Laurent, A.M., Site-specific retrotransposition of L1 elements within human alphoid satellite sequences, Genomics 46:130–131, 1997. The authors consider L1 insertion to be site-specific, requiring a sequence of 2–10 pyrimidines* followed by 3–7 purines*. This is at least true for insertion into (A+T)-rich sequences.
- Holmquist, G.P., Chromosome bands, Amer. J. Human Genetics 51:17–37, 1992.
- Jurka, J. and Kapitonov, V.V., Sectorial mutagenesis by transposable elements, Genetica 12:4–6, 2000.
- Wichman, H.A. et al., Transposable elements and the evolution of genome organization in mammals, Genetica 86:290, 1992.
- Usdin, K. and Furano, A.V., Insertion of L1 elements into sites that can form non-B DNA, J. Biological Chemistry 264:20742, 1989.
- Feng, Q. et al., Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition, Cell 87:907–913, 1996.
- Hamada, M., A newly isolated family of short interspersed repetitive elements (SINEs) in Coregonid fishes, Genetics 146:363–364, 1995.
- Kordis, D. and Gubensek, F., Horizontal SINE transfer between vertebrate classes, Nature Genetics 10:131–132, 1995. The element belongs to the LINE rather than SINE family. See also: Kordis, D. and Gubenesek, F., Unusual horizontal transfer of a long interspersed nuclear element between distant vertebrate classes, Proc. Nat. Acad. Sci. USA 95:10704–10709, 1998.
- Gojobori, T. et al., Patterns of nucleotide substitution in pseudogenes and functional genes, J. Molecular Evolution 18:367, 1982.
- Koop, B.F. et al., Primate Eta-globin DNA sequences and man’s place among the great apes, Nature 319:236, 1986.
- Li, W-H. et al., Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications, J. Molecular Evolution 21:58, 1984.
- Goldman, N., Statistical tests of models of DNA substitution, J. Molecular Evolution 36:189–190, 1993.
- Bull, J.J. et al., Exceptional convergent evolution in a virus, Genetics 147:1497–1507, 1997.
- Cunningham, C.W. et al., Parallel molecular evolution of deletions and nonsense mutations in Bacteriophage T7, Molecular Biology and Evolution 14(1):113–116, 1997.
- Broughton, R.E. et al., Conflicting phylogenetic patterns caused by molecular mechanisms in mitochrondrial DNA sequences, Systematic Biology 47:696–701, 1998.
- Graur, D. et al., Deletions in processed pseudogenes accumulate faster in rodents than in humans, J. Molecular Evolution 28:283, 1989.
- Shen M.R. et al., Evolution of the master Alu gene(s), J. Molecular Evolution 33:312, 1991.
- Jurka, J., Origin and evolution of Alu repetitive elements; in: Makalowski, Ref. 38, p. 33.
- Jurka, J. and Milosavljevic, A., Reconstruction and analysis of human Alu genes, J. Molecular Evolution 32:117–121, 1991.
- Britten, R.J., Quantitative study of Alu repeated sequences in primate genomes, in: Makalowski, Ref. 38, pp. 224–229, 1995. It is also interesting to note that deletions in Alus occur at nonrandom locations (p. 228). This further reduces the degrees of freedom by which one Alu repeat can diagnostically differ from another for purpose of orthologous matching.
- Furano, A.V., The biological properties and evolutionary dynamics of mammalian LINE-1 retrotransposons, Progress in Nucleic Research and Molecular Biology 64:282, 2000.
Support the creation/gospel message by donating or getting involved!

Answers in Genesis is an apologetics ministry, dedicated to helping Christians defend their faith and proclaim the good news of Jesus Christ.
- Customer Service 800.778.3390
- Available Monday–Friday | 9 AM–5 PM ET
- © 2026 Answers in Genesis