
Hemoglobin: An Exquisitely Designed, Multifunctional Protein
At a Glance
- Hemoglobin structure: The structure of hemoglobin is designed to carry out several different functions not for just transporting oxygen.
- Hemoglobin genes and their control: Genes which code for the subunits of hemoglobin are clustered on separate chromosomes; an α cluster and a β cluster. Expression of each gene in the cluster is coordinated throughout development; the expression of each gene is dependent upon its location in the cluster.
- Protection against superoxide radicals: Hypoxic (low oxygen levels) can lead to further tissue damage upon re-oxygenation through production of superoxide radicals by the mitochondria. Under such conditions hemoglobin and myoglobin can synthesize nitric oxide (NO) which increases O2 delivery by increasing blood flow and suppresses radical formation by the mitochondria.
- Cooperativity in oxygen (O2) binding and release: The subunit structure of hemoglobin permits more effective and efficient O2 loading and unloading.
- Binding of CO2, protons (H+) and other compounds: In addition to transporting O2, or holding it in reserve, hemoglobin also transports H+ and CO2 from the tissues to the lungs for release. Hemoglobin serves as an important intracellular pH buffer for red blood cells (RBCs).
- Hemoglobin as a sensor of metabolic demand for oxygen: The amount of O2 released at the tissues depends on several factors: the concentration of CO2, pH and pO2. All which exhibit changes dependent on the degree of metabolic activity.
- Fetal hemoglobin: A special hemoglobin synthesized by the developing baby that has a higher affinity for oxygen which allows it to withdraw O2 from the mother’s hemoglobin.
- Enzymatic activities, delivery of nitric oxide (NO) and protection against NO: Under normal conditions, when O2 is plentiful cytotoxic NO can be converted to non-toxic nitrates by a dioxygenase activity of hemoglobin. Under hypoxic conditions, hemoglobin can synthesize NO which can increase O2 delivery and suppress superoxide radical production by mitochondria.
- Matching hemoglobin structure with animal size: Animals of different sizes produce different hemoglobins that exhibit different sensitivities to the Bohr effect, which enables the coordination of O2 delivery with metabolic activity.
- Artificial, man-made hemoglobin: Several research programs have spent much money and time trying to develop an artificial hemoglobin that could be used in transfusions and other medical procedures. However, while much progress has been made, none are as efficient and effective at O2 delivery as the one designed by the Creator.
Introduction
Most multicellular organisms require oxygen (O2) in order to generate enough energy to sustain their life. We know that organisms are composed mostly of water. While this is essential for life, the solubility of O2 in water is too low to sustain life, and the diffusion of O2 in water is too slow. For these reasons it is necessary to have an efficient means of transporting and storing O2 for use by all the cells of an organism. Two proteins have been specifically designed to carry out these functions and more, hemoglobin (Hgb) and myoglobin (Mb). Hemoglobin transports O2 from place-to-place in the body via the blood, and Mb stores and transports O2 from place-to-place within a cell. Hemoglobin significantly increases the O2-carrying capacity of the blood by almost 70-fold! Human blood normally has an O2 concentration of ~200 mL O2/L of blood, plasma carries ~1.5% as dissolved O2 and Hgb 98.5%.
Most everyone has heard of Hgb; it is the red-pigmented protein found in red blood cells that gives blood its characteristic red color. The reason it is red is because it, like many proteins, has a heme group with an iron atom bound to it (fig. 1). The heme is a prosthetic group. A prosthetic group is a tightly bound, organic, non-protein portion of a protein. Prosthetic groups, when present, help a protein carry out its particular function(s).
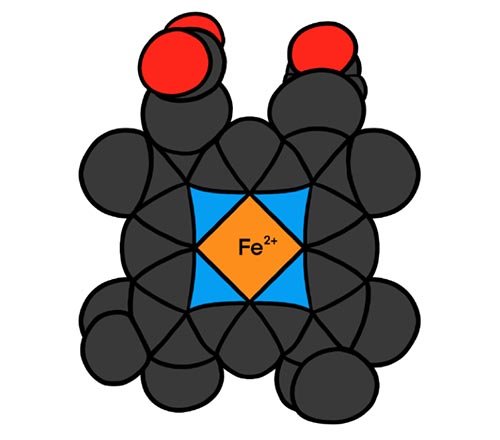
Figure 1. The heme prosthetic group, which binds O2 to the central Fe2+
We are all so familiar with the fact that blood transports O2 from the lungs to the rest of the body that we too often take it for granted.
We are all so familiar with the fact that blood transports O2 from the lungs to the rest of the body that we too often take it for granted. How many of us have stopped to think about exactly what is this Hgb? Does it do anything else besides carry O2? When we begin to examine life on the molecular level, we quickly begin to realize that Hgb, indeed all proteins, is specially designed with a purpose in mind; not just for one purpose, but for multiple purposes! We are left standing in awe of our Creator.
In this article we first examine the structure of Hgb and how this makes Hgb ideally suited as an O2 transporter. We’ll very briefly look at the genes that code for the various subunits of Hgb and how they are arranged on the chromosomes, and how their expression changes through development from embryo to adult, and how this is regulated. We will examine how hemoglobin structure enhances the binding and release of O2, making hemoglobin an exquisitely designed sensor of the metabolic demands for oxygen. We will make some comparisons between Hgb and myoglobin, another O2 binding protein found in muscle, and see how its structure suits it to be an excellent O2 storage protein in cells. We examine some associated enzymatic activities of Hgb and Mb that protect us from nitric oxide (NO), but at the same time provide a way to preserve and deliver NO to where it’s needed. We briefly look at Hgbs from other mammals and see that their Hgbs are matched to their metabolic demands. Lastly, we mention some research advances and shortcomings in attempts to make artificial Hgb to meet many medical needs.
Hemoglobin Structure
In adult humans, Hgb is composed of four separate polypeptide chains called subunits: two α and two β (sometimes two δ) subunits. Each α and each β combine to form an αβ-heterodimer. Thus, each Hgb molecule is made by combining two αβ-heterodimers (fig. 2). The dimers are typically designated as α1β1 and α2β2. In Hgb interactions between the α1 and β1 subunits and between the α2 and β2 subunits are relatively strong. The strong interactions within a dimer allow each dimer to move as a unit relative to the other dimer. The interactions at the interfaces between dimers; the α1-β2 and the α2-β1 subunits, however, are relatively weak. These weak interactions between dimers allows some freedom of movement which is essential for Hgb to carry out several of its functions (fig. 3).
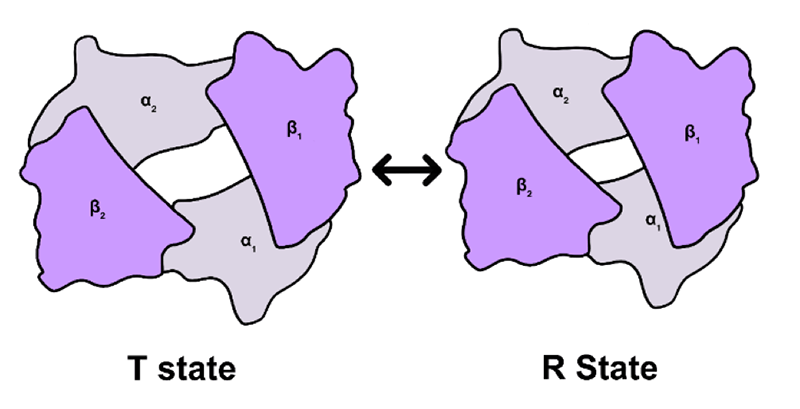
Figure 2. The hemoglobin molecule showing the two αβ-dimers. Depending on whether O2 is bound, the molecule can exist in either of two states: with O2 it is the R state and without O2 the T state.
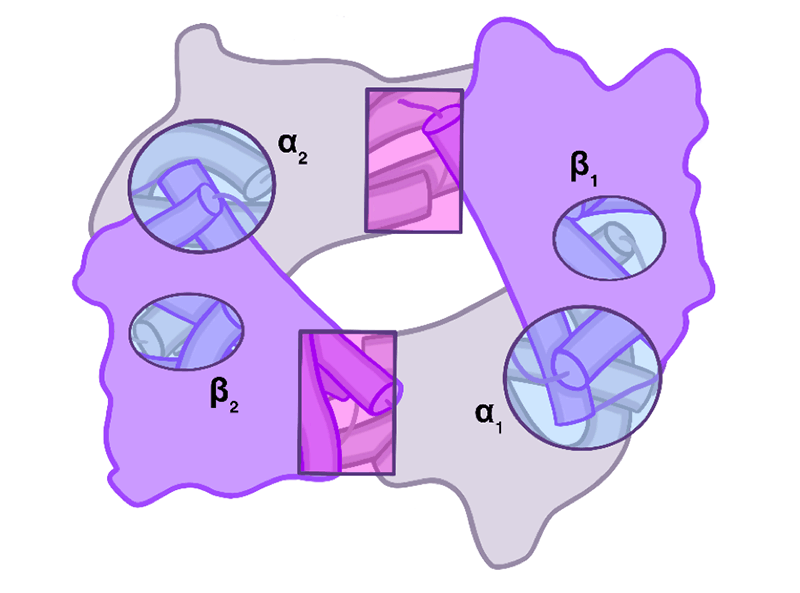
Figure 3. Circles and ovals are highlighting the relatively strong contacts between the α and β subunits of a dimer. The rectangular regions highlight the relatively weak sliding contacts between dimers. The sliding contacts permit rotation of one αβ-dimer relative to the other dimer. Movement at these latter contacts permits transition between the T-state and R-state of hemoglobin. This movement also leads to changes in size of the central cavity which affects binding of 2,3-bisphosphoglycerate (2,3-BPG).
Each α and β subunit contains a heme group with a bound iron atom. The iron atom is in the +2 oxidation state. Each iron can bind one O2, thus each Hgb molecule can bind up to four oxygen molecules. Myoglobin with its single polypeptide chain and single heme can bind only one O2 molecule.
When the heme group and its bound iron are separated from the globin (protein portion of Hgb) as in the lab, the iron is easily oxidized to the +3 oxidation state which cannot bind O2. Thus, it is essential that the iron in both Hgb and Mb be maintained in the +2 oxidation state. In the +3 oxidation state Hgb and Mb are called methemoglobin and metmyoglobin, respectively. Indeed, when tissues such as meat, which is high in Mb, and blood stand in the open air, they turn brownish as the iron in the Hgb and Mb becomes progressively oxidized to the +3 oxidation state. Hence the brown color of dried blood and aged meat.
How does the iron in Hgb or Mb remain in the +2 oxidation state when O2 is around? There are two factors that help to maintain the iron in the +2 state: (1) In both proteins the heme is non-covalently bound to the globin in a hydrophobic pocket; i.e., the amino acid side chains that make up the pocket are hydrophobic such as valine, isoleucine, phenylalanine these help to exclude water from the pocket; (2) A strategically placed histidine (His) amino acid, called the distal His, is located on the side of the heme that binds O2. Oxygen is more electronegative than iron so tends to withdraw electrons from the iron. This causes the iron to develop a partial +3 charge and the O2 to develop a partial -1 charge. The distal His forms a hydrogen bond with the O2 and stabilizes it (fig. 4). If the His was not there, the O2 would have a greater tendency to dissociate from the iron leaving it in the +3 state and unable to bind O2. The O2 leaving would leave as a superoxide radical (O2-•). Radicals are highly reactive and will destroy membranes, proteins and nucleic acids. Histidine is not the only amino acid capable of forming a hydrogen bond with O2, lysine and arginine can do the same. However, they are too big to fit into the O2-binding pocket as they would interfere with O2 binding. Only His has the right dimensions. Indeed, in most Hgbs studied a His is located in the same position relative to the heme. The evolutionist would say that the distal His has been “conserved” through evolution. However, the best explanation is that the distal His is a design feature. Virtually all multicellular organisms must have the ability to reversibly bind O2 and maintain the iron in the +2 oxidation state while transporting or storing it.
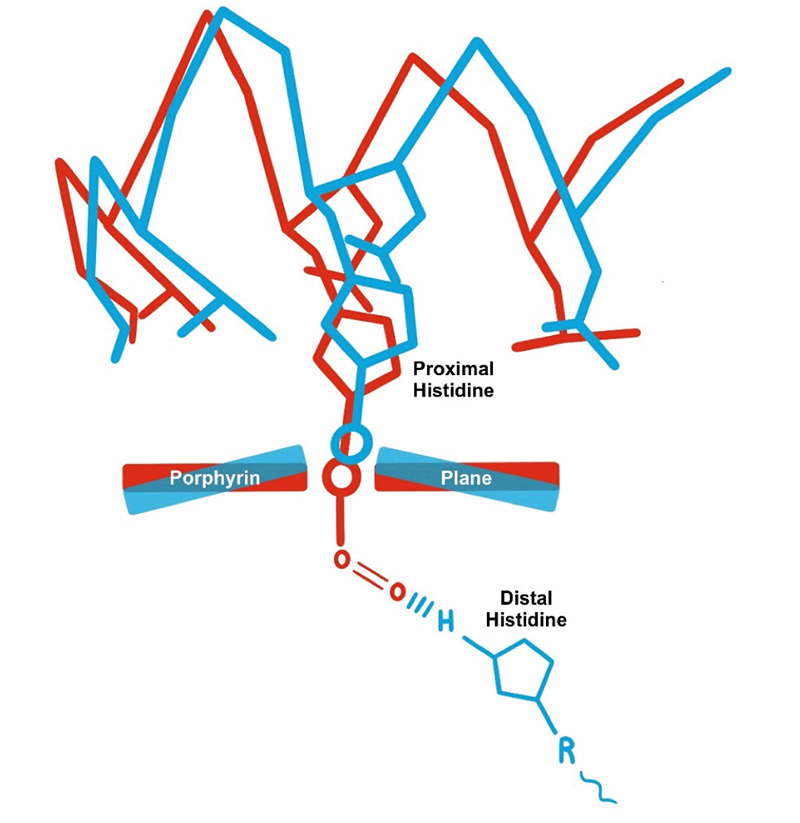
Figure 4. Shows several things: Changes in the heme, placement of the Fe2+, and changes in polypeptide chain near the proximal His upon binding O2. The distal His forms a hydrogen bond with the bound O2 to stabilize it and keep it from dissociating leaving the iron in the +3 oxidation state.
We live in a fallen world consequently O2 does dissociate from Hgb and Mb, leaving the iron in the +3 oxidation state. However, in God’s design red blood cells (RBCs) express the enzyme methemoglobin reductase that converts the iron back to a +2. Similarly, other tissues express a metmyoglobin reductase to convert Mb back to a +2 oxidation state.
Hemoglobin α and β chains and Mb have a very similar (almost identical) 3-dimensional, tertiary1, structure (fig. 5). This is interesting because only about 18% of their amino acids are identical! The best explanation is that the near-identical 3-dimensional shape is a design feature since the function of all three polypeptides is to bind to and transport O2. Thus, their function dictates their shape more than their amino acid composition, as would be expected from an evolutionary view. This is not unique to Hgb, indeed many other proteins that have similar functions have very different amino acid compositions but have very similar 3-dimensional conformations. In the evolutionary view, structure (or form) gives rise to function. However, this is better explained from a design perspective which suggests function comes BEFORE form; the form is designed to carry out the function. Indeed, is this not what we see and experience every day? All man-made devices, machines, or objects have their origins in the mind of a designer who designed their form to carry out particular functions.
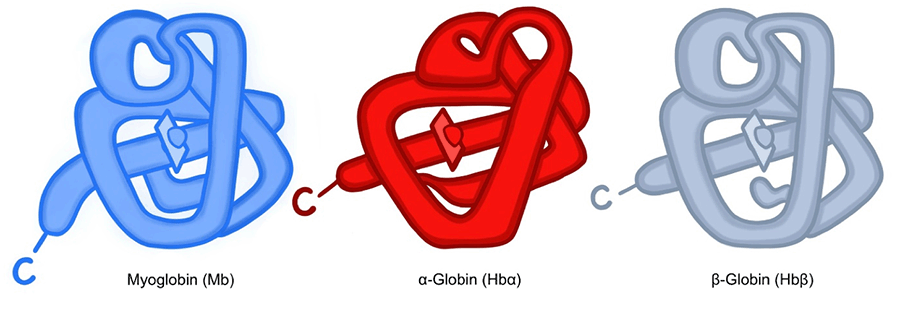
Figure 5. Note the similarity in 3-D structure between the single myoglobin molecule and the two subunits of hemoglobin. The similarity in structure is best explained by their similar function.
Before we continue our discussion of Hgb, let’s stop and remember that all proteins are composed of amino acids, but the proper sequence of amino acids is encoded in the language of nucleic acids, DNA. The Designer not only has a set of functions in mind for Hgb, but then must encapsulate this information in a “blueprint” to be followed whenever the subunits of Hgb are made. Each of the four subunits are made independently of each other, folded by yet other proteins known as chaperones, then the subunits self-assemble into the complete Hgb molecule. Exactly how blind chance could cause that to happen is a mystery.
Hemoglobin Gene Structure and Regulation
The Hgb molecule described above is that of adult human Hgb and is commonly designated as HgbA1. However, it is not the only Hgb produced by humans. There are three different embryonic Hgbs called Hb Gower 1, Hb Gower 2 and Hb Portland. There is a fetal Hgb designated HgbF. There is also another adult Hgb in the human population called HgbA2. It has two α chains and two δ chains which are similar to the β chains. Each of these is formed during different stages of development and are composed of different α-like and/or different β-like polypeptide chains. Many, perhaps most, of us know that RBCs are produced in the red bone marrow of many bones. But during development, red blood cells and their Hgbs are produced first in the fetal yolk sac, then the fetal liver, then the fetal spleen and finally the adult bone marrow (fig. 6). Thus, we see that there is a coordinated process of synthesizing different Hgbs by different organs during development. Each of these Hgbs is specifically designed to carry out the function of O2 transport under the various conditions that arise during development.
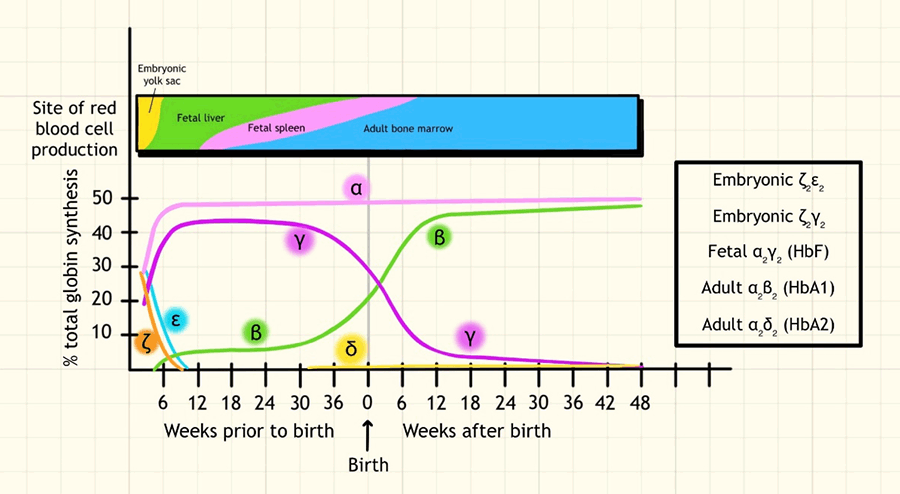
Figure 6. The various hemoglobins that are produced during development and after, their subunit composition and the tissues involved at the various times.
The α and α-like genes are located together on chromosome 16 and the β and β-like genes are located on chromosome 11 in humans. The gene for myoglobin resides on yet a different chromosome, number 22. Evolutionists believe that all the different Hgb genes and the one for myoglobin began as a single ancestral gene that through a series of gene duplication events followed by various mutations happening over hundreds of millions of years eventually gave rise to the genes of today. This scenario is pure speculation in that it was not observed, nor can it be repeated. Granted gene duplications and mutations can be explained by various known mechanisms, but this does not mean that this is how we got the genes for Hgb and myoglobin we have today. This becomes especially evident when we consider how these genes are arranged on the chromosome and how their expression is regulated (fig. 7). The β genes are arranged in a cluster, i.e., they are found next to each other in the same region of chromosome 11. Of particular interest is that the linear order of the genes on the chromosome is the same order in which they are expressed through development! That is, the first gene in the cluster is expressed first during development, the second gene is expressed second during development, and the last gene is expressed last during development. This has not been, nor can it be, explained by naturalistic mechanisms. Noted evolutionary geneticist, James Crow says:
“It is remarkable that in both the α and β group, the functional genes go from left to right in order of the time of their expression. Those at the left are expressed in the early embryo and the two on the right of each group (α1 and α2 and δ β) are adult hemoglobin components. The significance of the order is not known, but it seems too improbable to be a mere coincidence.” (Emphasis added)
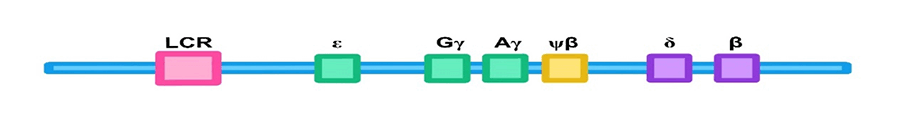
Figure 7. The β globin cluster on chromosome 11. The α globin genes are arranged in a similar cluster on chromosome 16. They also have a LCR (Locus Control Region) upstream of the genes.
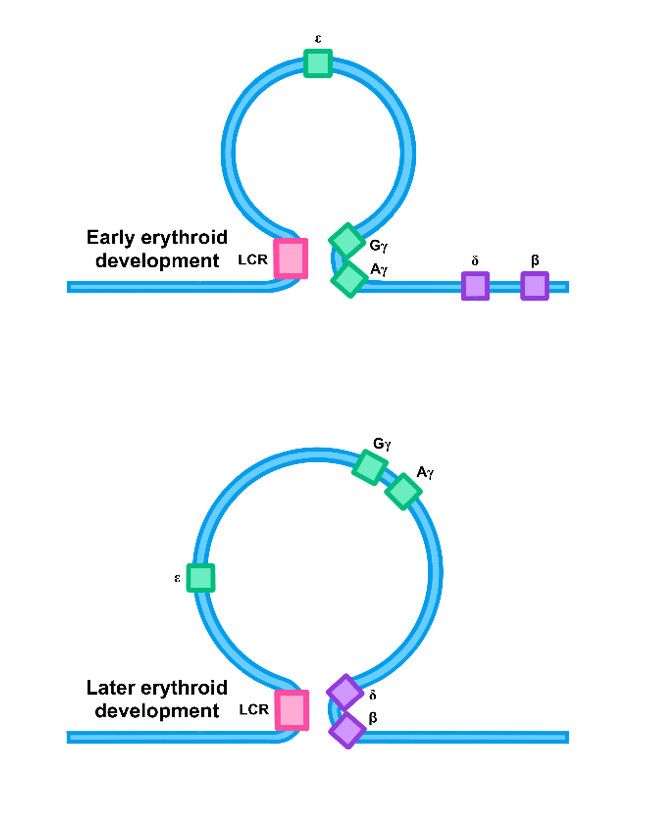
Figure 8. Regulation of the genes in the β-globin cluster. In very early erythroid development the ε gene was being expressed (not shown). Later in early development the expression pattern changes as the LCR and the appropriate genes are brought close together via looping of the DNA between them (top figure). At a still later time the γ genes cease being expressed and the adult β and δ genes are expressed.
Many, but not all, genes arranged in clusters are put under the regulation of a DNA sequence known as a Locus Control Region, or LCR. Proteins bound to this sequence regulate and coordinate the expression of the genes in the cluster. Exactly how this works is the subject of much research. Expression of the genes in both the α cluster and the β cluster is controlled by an LCR which resides upstream of the first gene in the cluster. It is thought that when the time comes for a gene to be expressed, the gene is brought close to the LCR by a DNA looping mechanism (fig. 8). Proteins bound at the LCR interact with proteins bound at the beginning of the gene (the promoter region) to aid in initiating transcription of that gene only. The proteins bound on the gene’s promoter and those at the LCR have to be designed to not only interact with the DNA at specific sequences, but also with each other in very precise ways. But that is a subject for another day.
Oxygen Binding
When RBCs are in the lungs, the Hgb adopts what is termed an R-state which has a high affinity for O2.
When RBCs are in the lungs, the Hgb adopts what is termed an R-state which has a high affinity for O2. The R-state is maintained by formation of specific weak bonds at the α1-β2 and the α2-β1 interfaces of the dimers.
Hemoglobin when binding O2, exhibits positive cooperativity; i.e., the binding of one O2 molecule to a subunit increases the affinity of binding a second O2 molecule to another subunit which, in turn, further increases binding of a third O2 and so on until the Hgb is saturated. This phenomenon is responsible for the S-shaped (sigmoidal) binding curve of Hgb (fig. 9). This graph is also known as the oxygen dissociation curve. Note the steep portion of the curve for Hgb. This shows that for slight increases in pO2 there is a significant rise in the percent saturation. Myoglobin has a rectangular hyperbolic dissociation curve indicating no cooperativity; consistent with the fact that it is a single polypeptide (fig. 10).
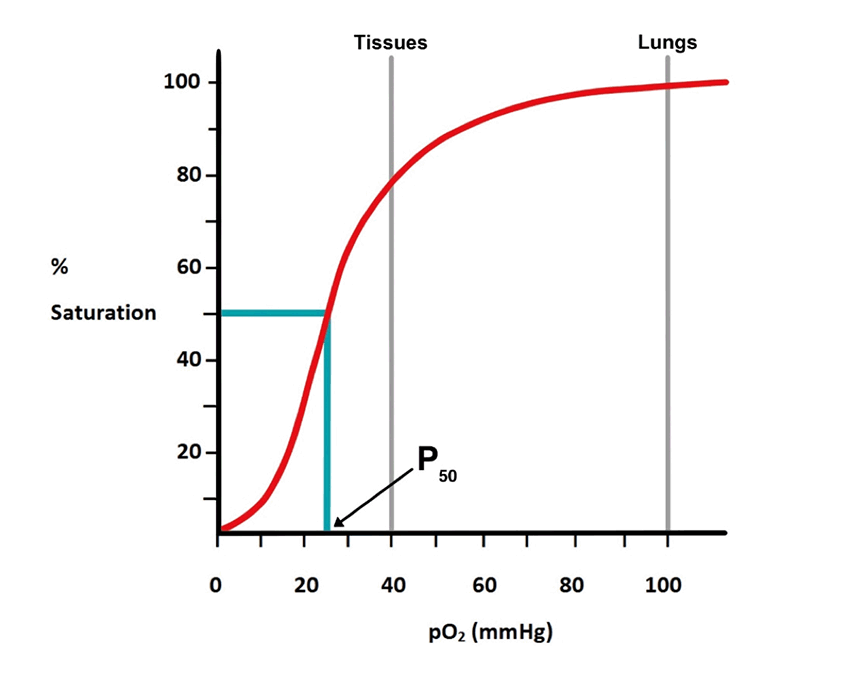
Figure 9. The sigmoidal shape of the hemoglobin curve is due to positive cooperativity between the subunits; the ability of one αβ-dimer to rotate approximately 15⁰ relative to the other αβ-dimer. The P50 is the pO2 that leads to 50% of hemoglobin molecules being saturated.
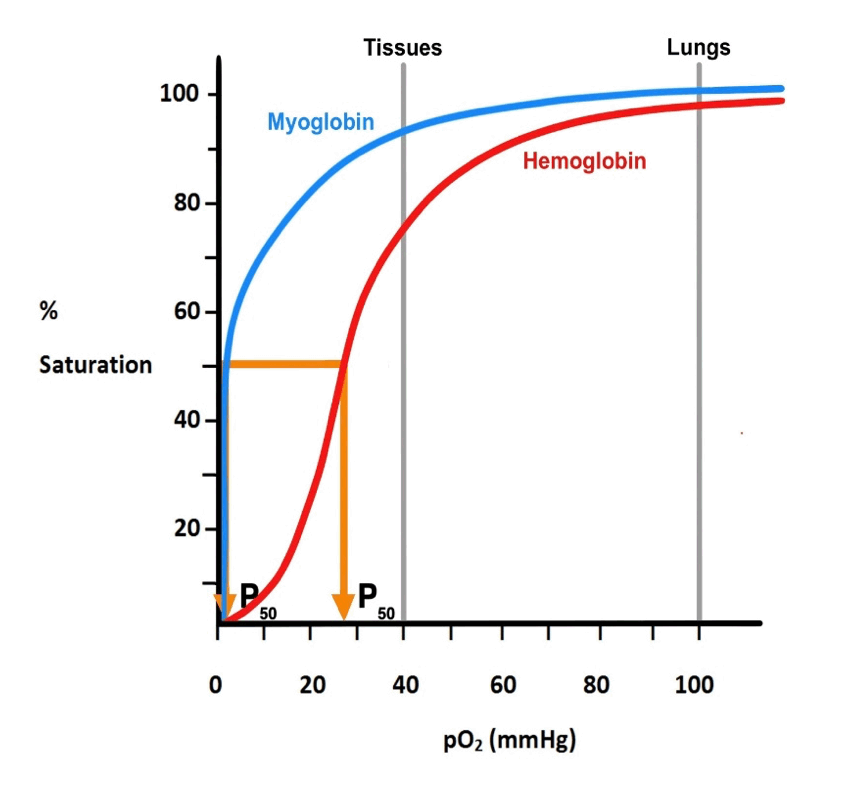
Figure 10. Saturation curves for hemoglobin and myoglobin. The Mb curve is hyperbolic indicating no cooperativity. This is consistent with the fact that Mb is a single polypeptide. It is also shifted far to the left of the Hgb curve indicating it has a much higher affinity for O2 than Hgb, also consistent with its function. The sigmoid curve of Hgb is indicative of positive cooperativity due to the subunit composition.
Positive cooperativity is explained as the binding of O2 to the iron in an α subunit of Hgb which causes the iron to move toward the heme slightly and the heme becomes a little more flattened (see FIGURE 4). The iron is attached to a His side chain, called the proximal His. The proximal His, in turn, is attached to adjacent amino acids in the chain thus pulling on the iron pulls on the His which pulls on the adjacent amino acids. This leads to a slight shift in all the parts of the α chain. Since that chain interacts through the weaker, sliding contacts with an adjacent β chain of the other dimer, this leads to slight shifts in the β chain such that O2 binds more readily to it. The shift in the β chain is then translated to other chains in the Hgb molecule such that each subunit makes the shift. This forms the oxy-Hgb, or R-state. With the binding of each O2, it becomes easier for the next one to bind. This is reflected in the steep up stroke section of the sigmoid curve. In the case of Hgb the last O2 binds with approximately 100X the affinity of the first O2! Thus, the shift from the deoxy, or T-state, to the oxy, or R-state, requires the entire molecule to move, hence weak interactions are required at the interfaces between the αβ dimers (fig. 3). Binding of O2 causes some bonds in the deoxy-T state to be broken and new bonds to be formed in the oxy-R state. Breaking of the bonds of the deoxy-T form allows one dimer to rotate about 15⁰ relative to the other.
The subunit structure of Hgb permits the positive cooperativity making it ideally suited for loading O2 in the lungs then unloading it at the tissues. Examine figure 10 and note that the Hgb dissociation curve, in addition to being sigmoidal, it is shifted to the right of the Mb curve. The reference point on each curve is termed the P50 which is defined as the amount of O2 (partial pressure of O2, pO2) that gives 50% saturation. Changes in the P50 are commonly used to express changes in O2 affinity; a higher P50 represents a lower affinity as it takes more O2 to reach 50% saturation, while a lower P50 represents a higher affinity. The right-ward shift of the Hgb curve shows that it has a higher P50 than Mb and thus a lower affinity for O2 (see fig. 10). The higher affinity (lower P50) of Mb for O2 makes it well suited for the capture of O2 from Hgb. In the lungs where pO2 is high, Hgb, in spite of its lower affinity, can easily load O2 (fig. 11).
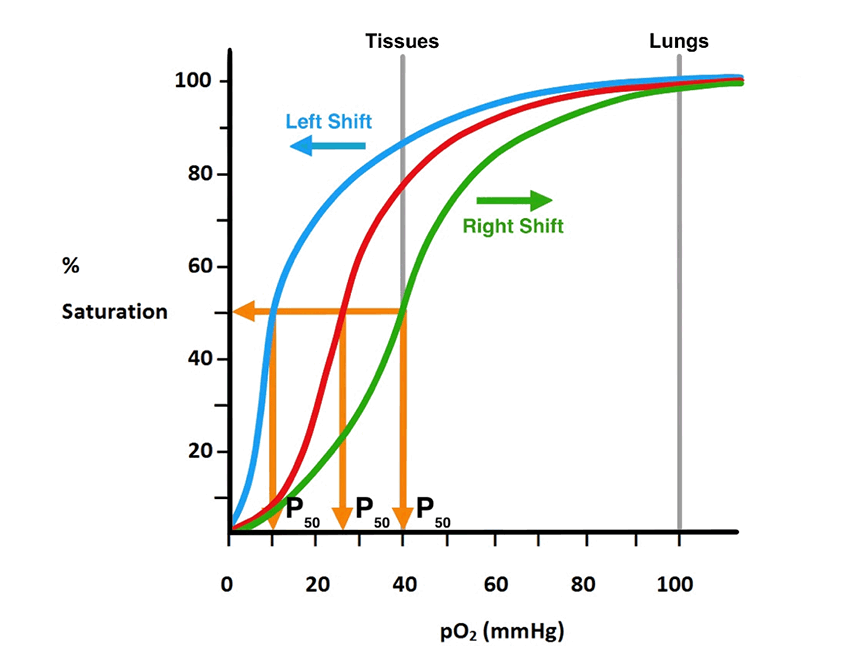
Figure 11. Shows how the P50, which reflects the O2 affinity of Hgb, changes with either a leftward or rightward shift in the dissociation curve. A rightward shift reflects a lower affinity. This is what happens at the tissue level. A leftward shift increases affinity. This is what happens in the lungs.
Oxygen Release and pH Buffering
In the tissues where pO2 is low, it is necessary to have another O2-binding protein that has a higher affinity for O2 than Hgb so that it may bind the O2 given off by Hgb and hold it in storage or shuttle it to the mitochondria where it is used in the generation of energy. This function is reserved for Mb.
As the blood circulates from the lungs where the pO2 is high to the tissues where the pO2 is low, it releases the O2. The lower affinity of Hgb relative to Mb helps to keep the Hgb from taking the O2 back.
Several factors induce a shift from the oxy-R state of Hgb in the lungs to the deoxy-T state in the tissues. These are an increase in temperature, an increase in CO2, a lowering of the pH (an increase in H+), and a metabolic intermediate produced by the RBC, 2,3-bisphosphoglycerate, or 2,3-BPG. As the blood circulates through metabolizing tissue, it encounters a slight increase in temperature. This is especially noted in very metabolically active tissues such as skeletal muscles during heavy exercise. This causes the O2 dissociation curve to shift right-ward indicating a lower affinity for O2. Carbon dioxide is a metabolic by-product released by most cells. The higher the metabolic rate the more CO2 is released into the blood. The CO2 diffuses into the RBC where an enzyme, carbonic anhydrase, acts on it to generate carbonic acid, H2CO3. This acid quickly deprotonates into H+ and HCO3- (bicarbonate). In the tissues the reaction proceeds from left to right with carbonic anhydrase catalyzing the first step.
CO2 + H2O ⇄ H2CO3 ⇄ H+ + HCO3-
The low pO2 of the tissues causes O2 to be unloaded from the Hgb. As O2 comes off, the Hgb binds H+ and CO2 at sites different from that of O2. Carbon dioxide binds to the Hgb molecule at the amino terminus of the four chains causing release of a H+. Hemoglobin binds the H+ released from CO2 binding and from the deprotonation of carbonic acid to help maintain the RBC pH within very narrow limits. Bound CO2 and H+ facilitate the shift of Hgb to the low-affinity, deoxy-T state. Binding of H+ to Hgb and inducing the R-to-T shift is known as the Bohr effect after Christian Bohr who first elucidated this phenomenon. This shift also causes the cavity between the subunits to enlarge enough so that 2,3-BPG can now bind in the cavity. All these actions stabilize the T-state so that Hgb cannot take back the released O2 (fig. 12).
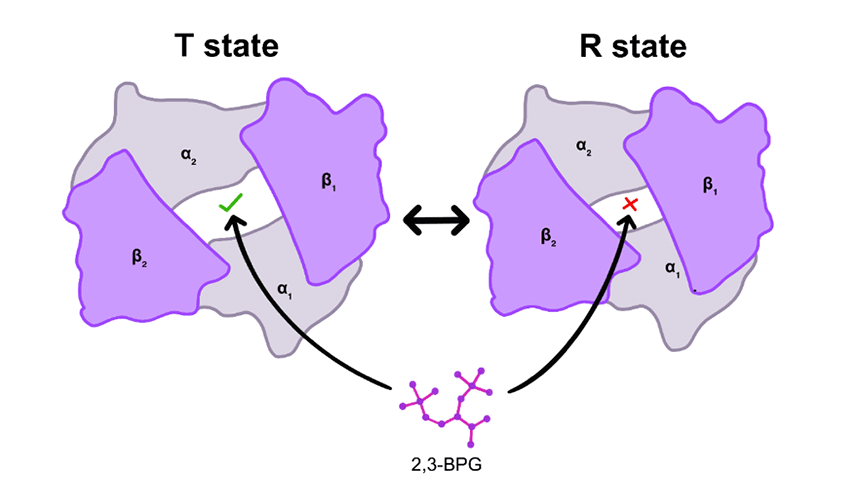
Figure 12. Binding of 2,3-bisphosphoglycerate (BPG) to the T state of Hgb only.
The T-state of Hgb is further stabilized by the binding of chloride ions (Cl-) from the plasma. The bicarbonate produced by deprotonation of H2CO3 would build up in the cell and cause the anhydrase reaction to go in reverse. To prevent this, the RBC has a transport protein in its cell membrane called the Band 3 protein (or bicarbonate:Cl- antitransporter) which transports one HCO3- out of the cell and one Cl- into the cell. The loss of a negative charge is balanced by the gain of one to maintain the proper membrane potential. The Cl- participates in a network of salt-bridges that help to further stabilize the T-state. Like 2,3-BPG, Cl- also binds to the central cavity and helps to reduce the mutual charge-charge repulsion of the positive charges.
When the blood returns to the lungs where the pO2 is much higher, all the actions described above are reversed; the carbonic anhydrase reaction proceeds from right to left, CO2 is generated and released due to the lower pCO2 in the lungs. It is then exhaled. The H+ on the Hgb are also released and used in the anhydrase reaction to make H2O. As the Hgb switches back to the oxy-R state with a lower P50 and higher affinity for O2, the Cl- comes off and is exchanged out of the cell for a bicarbonate ion. The bicarbonate ion is used up in the anhydrase reaction to generate CO2 exhaled. The Hgb O2-binding curve shifts back to the left (see fig. 11).
2,3-Bisphosphoglycerate (2,3-BPG) an Allosteric Modulator of Oxygen Binding
When one ascends to a higher altitude from sea-level for a period of time (days to weeks), the body acclimates in part by synthesizing more 2,3-BPG. This compound binds to and stabilizes the deoxy-T state of Hgb in order to facilitate greater O2 delivery to the tissues in spite of the lower pO2 of the higher altitude. The more 2,3-BPG the greater the rightward shift in the saturation curve thus leading to more O2 being released to the tissues (fig. 13). Those living at higher altitudes such as in Denver, CO, or Santa Fe, NM or in the Andes of SA, will have higher 2,3-BPG levels in their RBCs. Returning to sea-level causes a reduction in 2,3-BPG levels back to pre-acclimated levels. It takes about 1–1.5 days to make the adjustments in BPG levels.
Hemoglobin as a Sensor of Metabolic Demand for O2
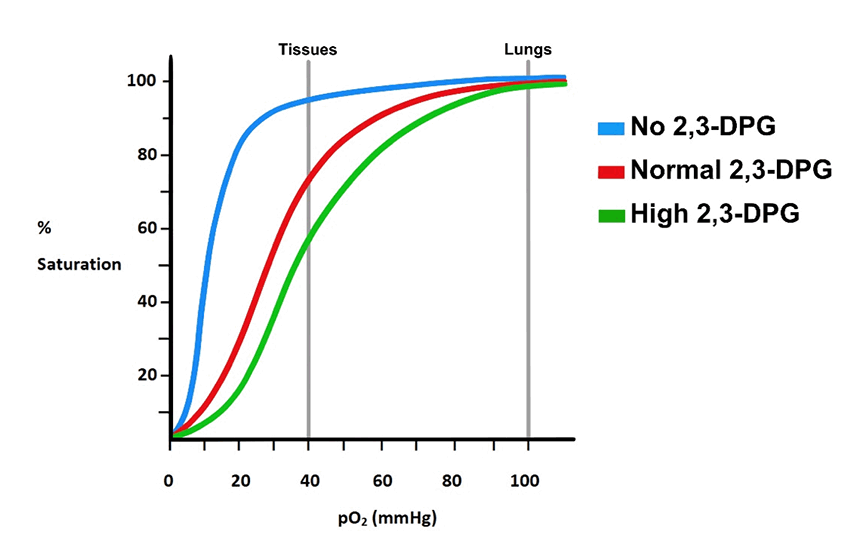
Figure 13. The effect of different concentrations of 2,3-BPG on the O2 dissociation curve. As can be seen, an increase in the BPG content of the RBC which happens at high altitudes causes a rightward shift in the curve to a lower affinity.
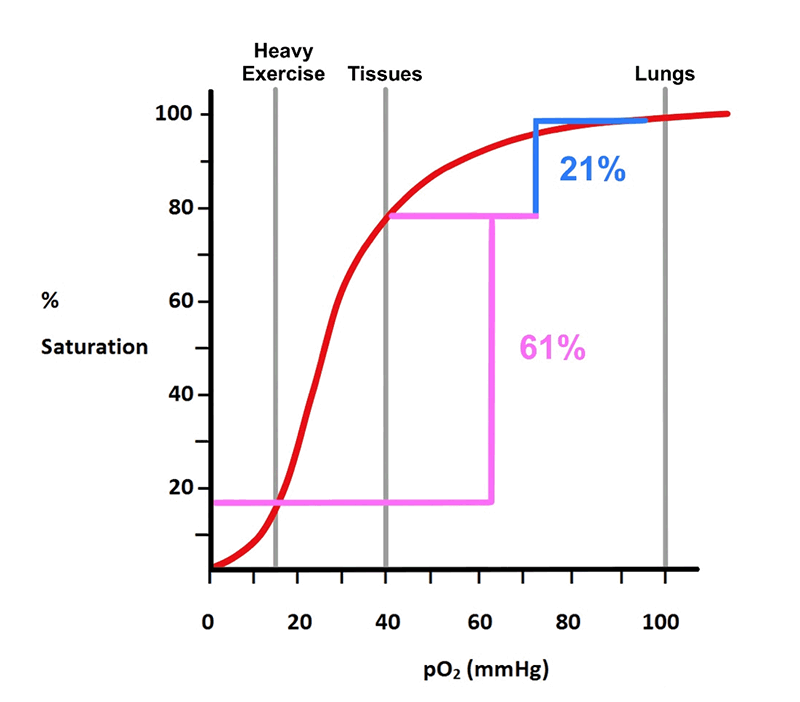
Figure 14. Shows that Hgb can release a significantly more O2 to the tissues if necessary.
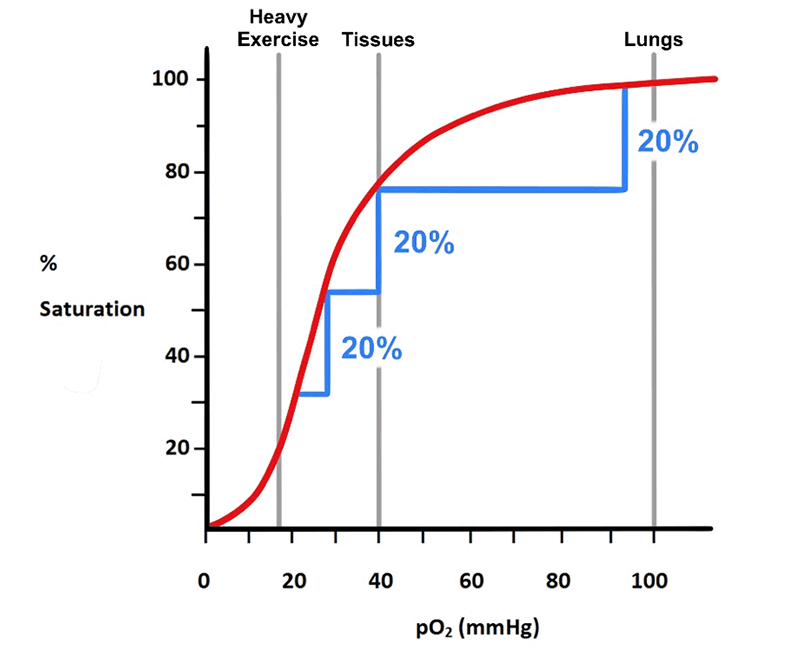
Figure 15. Shows that, in the steep portion of the curve, a small decrease on pO2 leads to release of more O2.
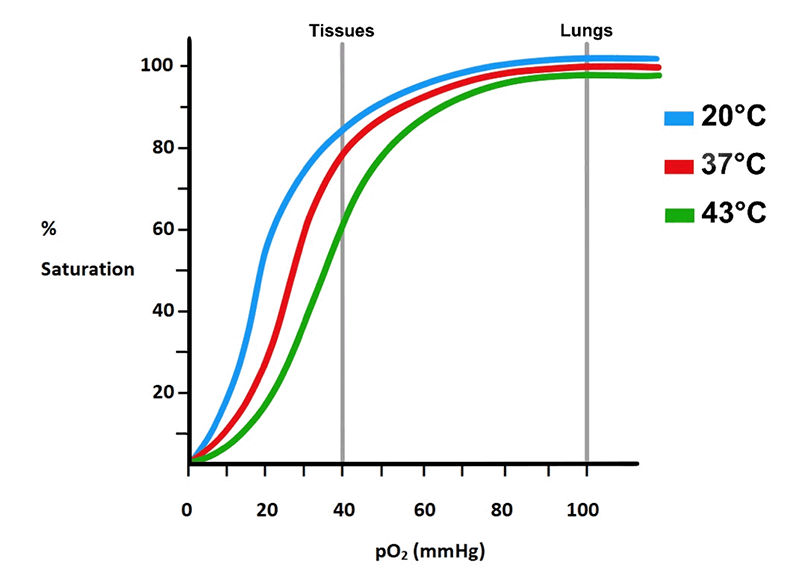
Figure 16. Effects of temperature on O2 affinity. Increases in temperature cause a rightward shift in the curve such that O2 affinity decreases. This is particularly important during higher metabolism, which causes the temperature in the tissues to rise.
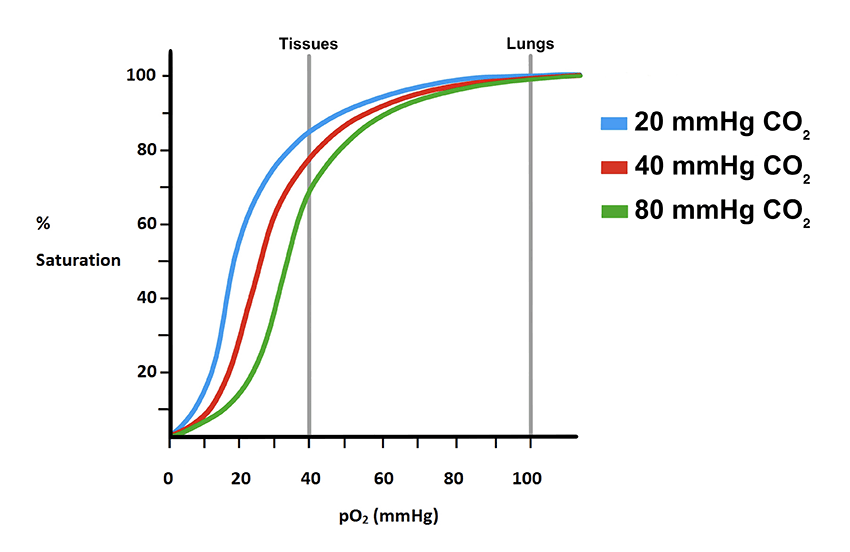
Figure 17. Effects of increasing CO2 on the saturation curve. As with increase in temperature and decreases in pH, increases in CO2 causes a rightward shift indicating a reduction in O2 affinity.
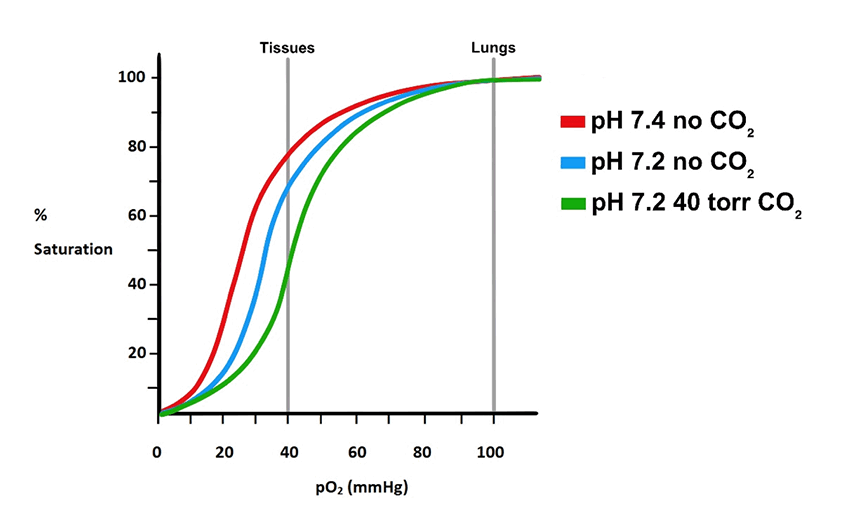
Figure 18. As pH decreases, the curve shifts rightward. If there is also an increase in CO2, there is a further rightward shift further decreasing the O2 affinity.
For all air-breathing organisms, adequate amounts of O2 delivery to the tissues is essential to life as it is used in the generation of energy from the mitochondria. During times of relative rest, metabolism ‘runs’ at a pace capable of keeping up with the energy demands of the tissues. Hemoglobin, under these conditions, releases about 21% of its O2 to the tissues; when it returns to the lungs, it still is about 79% saturated. However, should energy demands increase as in fight-or-flight situations, Hgb is capable of releasing much more O2, about 61% or more (fig. 14). We see then the structure of Hgb is such that it has the capability of releasing a little O2 or a lot of O2 depending on the energy demands of the tissue at any given moment; it can hold O2 in reserve.
Examining the steep region of the Hgb O2-binding curve shows that for even a slight decrease in pO2 will cause the unloading of significantly more O2 (fig. 15). Again, this could only be achieved by a protein with subunits that interact the way those of Hgb do.
In a very real way Hgb acts as a sensor of the energy, and thus O2, demands of the body. As described above, the dissociation curve shifts to the right when Hgb encounters a region of slightly elevated temperature (fig. 16). If the pCO2 is also elevated, as is usually the case, the curve can shift further to the right (fig. 17). If the pH is also slightly lower, the curve will shift even further to the right. Each shift to the right allows Hgb to release more O2 (fig. 18).
Delivery of Nitric Oxide and the Enzymatic Activities of Hemoglobin and Myoglobin
Nitric oxide (NO) is a gas that acts also as a neurotransmitter in the nervous system and as a second messenger in various signaling systems. Red blood cells make up the body’s largest intravascular reservoir of NO, and Hgb is indispensable to its formation, storage and release. In the vascular system NO is an important potent local vasodilator synthesized by the endothelium of arterioles. It binds to its receptor on smooth muscle cells surrounding arterioles and brings about relaxation allowing greater blood flow. This facilitates the transfer of O2 from Hgb to the tissues. Nitric oxide has a very short half-life (1–5 seconds) because it is destroyed rapidly. However, its lifespan can be greatly increased minutes to hours by binding it to Hgb and/or small sulfur-containing compounds in the blood such as glutathione. It is also a free radical and thus potentially toxic to cells.
Nitric oxide binds to the iron in the heme of Hgb with much greater affinity than O2 (approximately 10,000X greater!). Nitric oxide can form a stable bond to the heme-iron only when Hgb is in the deoxy-T state because in the oxy-R state NO could be rapidly oxidized to NO3-. The interaction between Hgb and NO is, then, controlled by the transition of Hgb from the oxy-R state to the deoxy-T state. When in the tissues, as O2 is released it leaves the heme-iron available to bind NO. A strategically placed Cys faces the surface of Hgb where it can easily transfer a bound NO to glutathione for delivery to the smooth muscle. Returning to the lungs O2 begins to bind to Hgb and CO2 and H+ leave causing it to convert to the oxy-R state. This shift now places the crucial Cys close to the heme-iron. At this point the NO on the heme-iron is transferred to the Cys. This leaves the heme-iron vacant to accept an O2. Upon return to the tissues O2 is released and Hgb converts back to the deoxy-T state. The Cys with its bound NO is back on the surface and NO is released to certain membrane proteins of the RBC where it is then transferred to glutathione for transfer to the smooth muscle cells. Thus, we see that the binding, transport and release of NO is dependent on the ability of Hgb to transition between the R- and T-states. A transition only made possible by the specifically designed subunit structure of Hgb.
In addition to its ability to transport O2 from the lungs to the tissues and H+ and CO2 from the tissues to the lungs, Hgb has two often overlooked enzymatic activities: nitric oxide dioxygenase and nitrite reductase. These two activities are, however, dependent upon whether the molecule is in the R- or T-state. Thus, these activities depend on the ability of Hgb to change its conformation. Under normal conditions when O2 is plentiful, oxygenated Hgb, in the R state, acts as a nitric oxide dioxygenase by adding its bound O2 to the NO to generate a non-toxic nitrate ion (NO3-). In the process, however, methemoglobin is generated, but as stated above methemoglobin reductase acts to regenerate the +2 oxidation state.
Under hypoxic (low O2) conditions Hgb, along with Mb, plays a protective role in suppressing the production of superoxide radicals by the mitochondria. Tissues subjected to hypoxic conditions suffer from greater tissue damage when O2 levels are restored. Under these conditions, mitochondria generate more superoxide radicals than normal. Hemoglobin exhibits the nitrite reductase activity only when in the deoxy-T state. The reductase activity synthesizes NO from ever-present nitrite ions (NO2-). The new NO increases O2 delivery by causing relaxation of the smooth muscle cells of arterioles and increasing blood flow and it also acts to suppress the production of radicals by the electron transport chain in the mitochondria thus reducing tissue damage when O2 delivery is resumed.
Fetal Hemoglobin, HgbF
This too is clearly a design feature that cannot be derived through the trial-and-error method of evolution.
Fetal Hgb also has α chains, but instead of the adult β chains, it has two γ chains which have a different amino acid composition. The central cavity in HgbA1 is lined with positive charges to interact with the negatively charged 2,3-BPG (fig. 19). Fetal Hgb does not have as many of these positive charges and thus cannot bind 2,3-BPG nearly as well. This means that HgbF has a higher affinity for O2 (lower P50) than the adult Hgbs (fig. 20). This makes sense when one considers that the baby must take its O2 from the mother’s blood. This too is clearly a design feature that cannot be derived through the trial-and-error method of evolution. All animals whose young develop inside the mother’s body require an effective and efficient method of getting O2 from the mother’s blood would have to have this feature. This is just one of the many changes that would have to take place and work perfectly from the get-go if transitioning from an egg-laying animal to one that engaged in live-birth such as mammals. There is no satisfactory evolutionary explanation for how this could have happened, only that “it evolved”. Virtually all evolutionary explanations of anything complex amounts to nothing more than simply “evolution did it”; evolution is invoked as the materialists “God of the Gaps”.
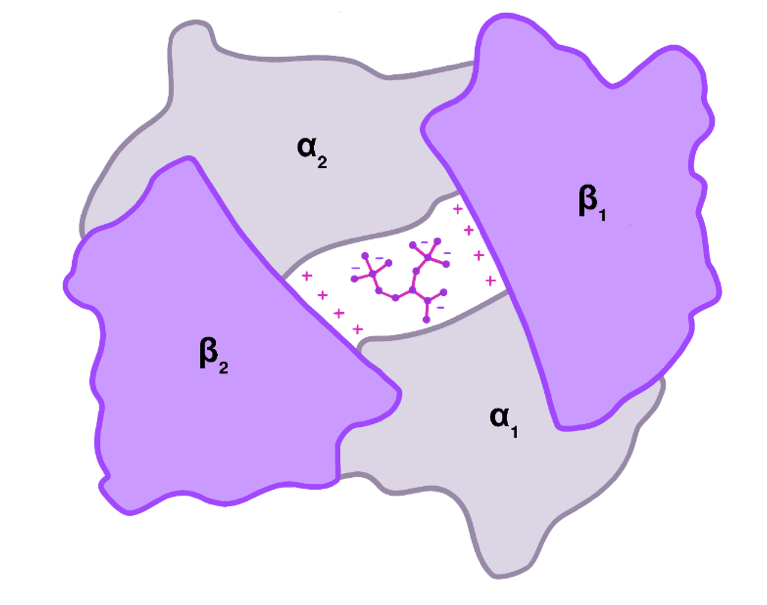
Figure 19. Binding of 2,3-BPG to the central cavity of Hgb in the T state. Positive charges on the β subunits interact electrostatically with the negatively charged 2,3-BPG. Fetal Hgb has γ subunits instead of the β subunits. The γ subunits do not have the positive charges and consequently do not bind 2,3-BPG.
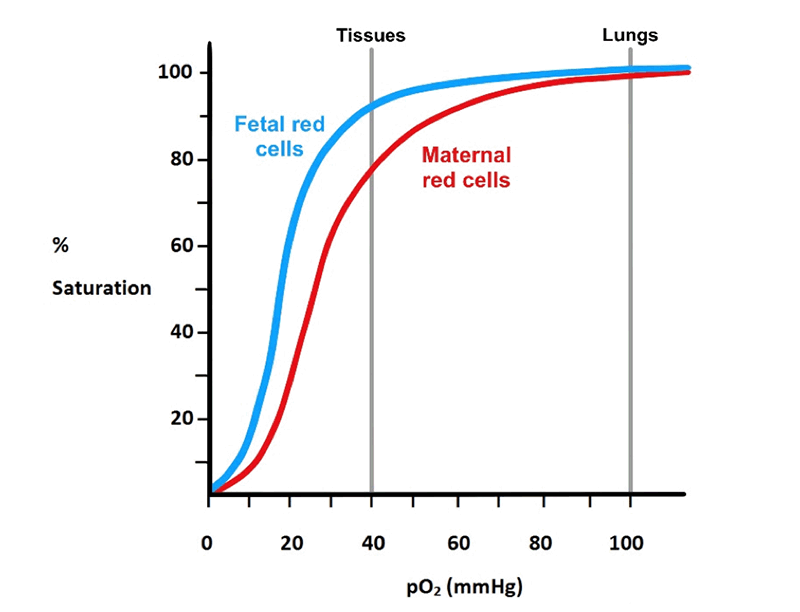
Figure 20. Shows the leftward shift of fetal Hgb dissociation curve indicating a higher O2 affinity than that of maternal Hgb. The higher affinity of fetal Hgb allows it to “pull” O2 from maternal blood.
Hemoglobin of Other Animals
The Hgb of other mammals have been well studied. It is interesting that there is a correlation between the size of the animal and the O2 affinity of the Hgb. Smaller animals such as mice have high metabolic rates and thus require much more O2 whereas larger animals such as an elephant have much lower metabolic rates and don’t require as much O2. Examination of the Hgb amino acid compositions of a number of mammals ranging in size from a mouse to an elephant reveals the different amino acid compositions of the animals are such that the O2 affinities of their respective Hgb are commensurate with their metabolic activities; larger animals with lower metabolic activities have Hgb with higher affinity and smaller, high metabolic animals, have a Hgb with lower affinities. These differences are attributable to differences in the sensitivity of the Bohr effect.
Artificial Hemoglobin
To make an artificial hemoglobin that can do all that our natural protein does and with the same efficiency requires a lot of intelligent input.
Much time and money has been, and is being, spent in search of an artificial Hgb that doesn’t have the problems presented by using human blood. Unfortunately, all efforts have been plagued with various problems such as too high or too low affinity for O2, binding NO too much so as to cause greater vascular resistance, causing an immune reaction, not able to also reversibly carry and transport CO2 and H+, and not able to release O2 in accordance with the metabolic demands of the tissue. Some new approaches now focus on using the natural Hgb enclosed in liposomes or nanoparticles acting as artificial cells.
That said, research to find a suitable hemoglobin substitute should continue. However, thus far all the research shows that to make an artificial hemoglobin that can do all that our natural protein does and with the same efficiency requires a lot of intelligent input. Too many are the factors that must be considered in designing such a protein that it is beyond credulity to accept that a blind, unthinking process such as evolution could do it.
Summary
In this article we have taken a little closer look at but one of the many thousands of proteins designed by our Creator. We see that Hgb does much more than just transport O2 from the lungs to the tissues. It has a structure that allows it to also bind CO2 and H+ thus acting as an important intracellular pH buffer in the RBCs. Its structure enables it to reversibly bind O2 with greater efficiency and effectively deliver O2 to meet the ever-changing metabolic demands for O2. The arrangement and coordinated expression of the genes which encode the subunits defy naturalistic explanations. Truly, Hgb exhibits all the hallmarks of design! It is an exquisitely designed, multifunctional protein.
References
Baynes, John W. and Marek H. Dominiczak. Medical Biochemistry, 5th ed. Amsterdam: Elsevier Publishing, 2019.
Crow, James. Genetics Notes, 8th ed., 170. New York: Macmillan Publishing, 1983.
Devlin, Thomas M., editor. Textbook of Biochemistry: With Clinical Correlations, 7th ed. Hoboken, NJ: John Wiley & Sons, Inc., 2011.
Garrett, Reginald H. and Charles M. Grisham. Biochemistry, 6th ed. Independence, KY: Cengage Learning Publishin, 2017.
Christopher K. Matthews, Kensal E. van Holde, Dean R. Appling and Spencer J. Anthony-Cahill. Biochemistry, 4th ed. London: Pearson Publishing, 2013.
Schmidt-Nielsen, Knut, Animal Physiology, 5th ed. Cambridge: Cambridge University Press, 1997.
Voet, Donald and Judith Voet. Biochemistry, 4th ed. Hoboken, NJ: John Wiley & Sons, Inc., 2011.
Answers in Depth
2020 Volume 15
Answers in Depth explores the biblical worldview in addressing modern scientific research, history, current events, popular media, theology, and much more.
Browse VolumeFootnotes
- Protein structure is hierarchical. There are four levels of structure: (1) Primary structure is the amino acid sequence; (2) Secondary structure is the specific 3-dimensional structure formed by a particular amino acid sequence. Certain amino acid sequences have a tendency to form either a helix, called an α-helix, or a sheet-like structure, called a β-pleated sheet; (3) Tertiary structure describes the particular folding of the helices and β sheets in 3-dimensions; (4) Quaternary structure is the combination of two or more polypeptides each folded into its own 3D shape. All polypeptides have a tertiary structure. Hemoglobin has a quaternary structure in that it is composed of four separate polypeptides.
Support the creation/gospel message by donating or getting involved!

Answers in Genesis is an apologetics ministry, dedicated to helping Christians defend their faith and proclaim the good news of Jesus Christ.
- Customer Service 800.778.3390
- Available Monday–Friday | 9 AM–5 PM ET
- © 2026 Answers in Genesis