The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
Claims about high genomic DNA sequence similarity between humans and chimpanzees are typically made to audiences that do not understand the various layers of technology and ideological bias imposed upon the origination of the data in question. The recent human-chimp Y-chromosome project introduced a number of important genomic tools to achieve a considerably less-biased analysis. The results indicated a much higher level of dissimilarity in both gene content and overall sequence similarity than the previously reported levels up to 99% similarity. As of yet, no similar study utilizing a less-biased genomic framework for autosomal regions has been reported. When evaluating comparisons between genomes using DNA sequence, it is important to understand the nature of how that sequence was obtained and bioinformatically manipulated before drawing any conclusions. It is not uncommon to arrange the sequence of a genome for which little is known by using the genome of a hypothetical closely related organism that has better developed genomic resources. It is also not uncommon to first screen the framework model genome to find regions of high similarity prior to any comparative analyses and to even omit gaps in the final DNA alignments before determining sequence identity. As a result, evolutionary bias literally colors every aspect of the DNA analysis and annotation. Understanding the technology used to produce a comparative genomic product for inter-genome studies is required prior to making any definitive conclusions about the data presented. At present, a considerably more unbiased approach to comparative genomics needs to be applied to the analysis and annotation of genome.
Keywords: comparative genomics, human-chimp similarity, human genome, chimpanzee genome, DNA sequencing, genome sequencing, cloning DNA
Introduction
The ability to sequence the DNA of an organism’s genome was an important scientific advance that radically changed many aspects of molecular biology and genetics in both the academic and private sectors. Unfortunately, many discussions and interpretations surrounding genomic sequence, particularly those of a comparative nature, are errant or misleading because of the type of DNA sequence in question. Depending on the type of research approach and technologies used to produce the overall DNA sequence assembly for a particular organism, certain limitations to its application and usage must be taken into account when applying it for any comparative purpose.
Not surprisingly, the role of available research funds weighed against the cost per base of DNA sequence is, in most cases, the deciding factor on the overall amount and quality of sequence produced. Getting more “bang for the buck” is generally the way grant funds are used when it comes to DNA sequencing. This general ideology is true of many post-human genome research projects which incorporate a DNA sequencing strategy called “whole genome shotgun sequencing”. This type of technology takes on particular significance when taking into account the massive amounts of data now being produced using next generation “massively parallel” sequencing technologies.
In 2004, the human genome was formally completed in regard to sequencing the major euchromatic sections (International Human Genome Sequencing Consortium 2004). In 2005 (The Chimpanzee Sequencing and Analysis Consortium), a rough draft of the chimpanzee genome was reported with the hope that its availability would vindicate the claims of biologists who had been promoting high similarity (95% or greater; Britten 2002) associated with an ape to human evolutionary transition. Years before the DNA revolution began, chimpanzees were often positioned in the evolutionary tree closest to humans out of all the extant apes. Some biologists even went so far as to say that humans and chimps should be placed in the same genus and considered separate species (Wildman et al. 2003). However, most scientists recognized the vast behavioral and anatomical differences that exist between humans and chimps and do not agree that they should be placed in the same genus (Taylor 2009). In addition, recent research has shown that some sections of the human genome are more similar to orangutan, and not chimpanzee producing evolutionary aberrant DNA patterns called “incomplete lineage sorting” (Hobolth et al. 2011).
Brief History of DNA Sequencing Technology
To fully understand the ramifications of the incredibly large amount of DNA sequence data currently available today in the world’s public repositories, it is important to first take a brief look at the history of DNA sequencing technologies. This will help explain why certain approaches were taken to sequence certain organisms and also allows an understanding of the resulting overall quality and usability for that particular sequence set. For a time-line of selected major events in the history of DNA sequencing research related to sequencing, see Fig. 1.
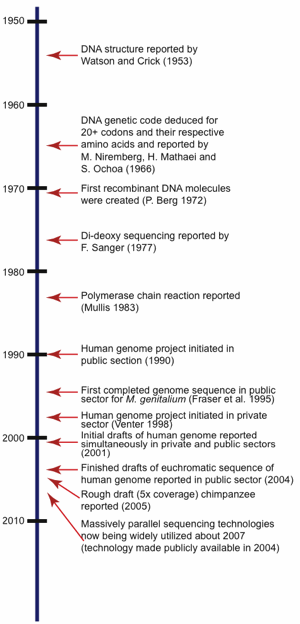
Fig. 1. Timeline showing significant milestones related to the history of DNA sequencing.
The whole modern phenomenon of DNA sequencing was introduced by the work of biologist and chemist Fred Sanger (Sanger, Nicklen, and Coulson 1977), research that earned him the Nobel Prize. Surprisingly, the basic chemistry invented by Fred Sanger, referred to as Sanger-style sequencing, has remained essentially the same from its earliest years until the present time. Drastic improvements in Sanger-style DNA sequencing since 1977 were largely achieved through four areas:
- the introduction of the polymerase chain reaction (PCR) and in improvements in the basic chemical components (various enzymes, reagents and DNA fragment labeling),
- the automation of sample preparation via large-scale microtiter plate (primarily 96 and 384-well formats) systems using robotically automated pipetting and thermo-cycler platforms,
- automated laser-based fragment detection systems which evolved from 96-lane slab gel systems to extremely high-throughput/automated robotic platforms using large arrays of individual capillaries that could resolve DNA fragments in 96 or more sequencing reactions in a matter of just a couple of hours; and then automatically reload themselves, and
- bioinformatic and computational advances in hardware and software to edit, process, and submit massive amounts of DNA sequence data to both local and off-site database repositories. Advances in laboratory information management systems (LIMS) contributed to the overall automation and integration of the overall process.
One important feature of modern Sanger-style sequencing is the long high-quality read lengths that can be achieved. Under relatively optimal conditions, high-quality DNA sequence with a rate of only 1 error in 10,000 bases can be routinely obtained with average individual read lengths up to ~1,200 bases. The public human genome project was largely completed using Sanger-style technology on DNA libraries constructed from mapped large-insert DNA clones (International Human Genome Sequencing Consortium 2001, 2004). Slab-gel DNA sequencers were used at the beginning of the project and were eventually replaced with first-generation capillary technology.
Currently, next generation DNA sequencing technologies based on an overall strategy called massively parallel sequencing (Mardis 2008; Rogers and Venter 2005), have increased overall total DNA sequence output. However, one inherent drawback to massively parallel sequencing as a whole is the dramatic reduction in the amount of high quality sequence per individual read. Based on the next generation technology variant, individual read lengths vary from about 25 bases to 100 bases (Mardis 2008) with some recent claims by machine suppliers as high as 400. The overall trend is that the more bulk sequence produced by a particular technology within a certain span of time, the shorter the average read length of the individual sequences. Massively parallel sequencing has important ramifications for comparative genomics that will be discussed after some background information on genome sequencing strategies is discussed.
Approaches To Genome Sequencing
The first genomes sequenced were small and microbial in nature and included several species of bacteria (Fraser et al. 1995; Mushegian and Koonin 1996). This is because the DNA in bacterial genomes is relatively void of non-protein coding DNA sequence which is often repetitive and difficult to sequence and computationally assemble. With highly repetitive genome sequence in higher eukaryotes, certain blocks of DNA sequence are repeated for very long stretches. The problem in such cases is not that the chemistry is unable to sequence the DNA, but the computational assembly of the repetitive sequence reads to form a single long error-free contiguous DNA sequence (contig) is confounded. In addition to the computational limitations of assembling highly repetitive sequences, the incorporation of a single errant sequence into a contig can also pull in a large number of other related errant sequences, producing sequencing chimeras. To solve this problem, techniques to jump over these areas of the genome using various types of frameworks and bridging scaffolds were implemented. Nevertheless, genome sequencing first tested the waters with small non-repetitive genomes that were easily assembled and then moved on to some of the more challenging eukaryotic genomes such as fruit fly, nematode, and human.
Genetic Maps
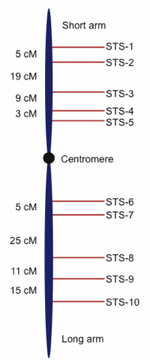
Fig. 2. Hypothetical genetic map showing sequence tagged sites (STS) or genetic markers with recombination-based distances between them demarcated in centimorgans (cM, also referred to as map units). Genetic marker nomenclature is diverse; the STS usage in this figure is for illustration purposes.
For the public human genome project, as well as several other initial eukaryotic genomes such as nematode and fruit fly, a frame-work based approach was developed to methodically sequence the genomes. In a framework approach, a variety of genomic tools are integrated to first form a genomic scaffold that can be used to identify targeted regions to sequence in addition to arranging and orienting sequencing reads (Meyers, Scalabrin, and Morgante 2004; Warren et al. 2006). The first part of the scaffold is called a molecular genetic map, which involves the placement of DNA landmarks throughout the genome by observing how DNA markers segregate in the offspring of controlled matings or in the case of humans, utilizing the extant pedigrees of large families (Kong et al. 2002).
Genetic mapping projects produce hundreds to thousands of DNA markers positioned in the proper order along chromosomes and separated by relative frequency-based distances called centimorgans. Without going into any more detail than this, it is sufficient to note that the process of genetic mapping can produce a rather detailed map of a genome that shows specific landmarks along chromosomes, much like a roadmap shows cities positioned along a highway (see Fig. 2 for an example of a genetic map). While genetic maps can be rather detailed, the distance between landmarks is not a physical distance that can be measured in actual base pairs of DNA, but rather represents a centimorgan unit which is a relative distance based on frequency of recombination between linked chromosomal sites.
Physical (Contig-Based) Clone Maps
The second key component of a genomic framework is a physical map, often referred to as a contig-based clone map which provides literal physical distances between points in the genome (Meyers, Scalabrin, and Morgante 2004; Warren et al. 2006). Cloning DNA fragments was a technology first developed in the early 1970s shortly after the discovery of restriction enzymes; proteins that cut DNA at specific sequence sites. In cloning DNA, the restriction fragments of the target organism’s DNA are placed in a small piece of engineered circular DNA called a plasmid. These plasmids are then transferred into lab strains of E. coli where they are maintained, replicated, and frozen for storage. The cloned DNA can be placed in arrayed sets of clones in microtiter plates called libraries.
These libraries are often frozen at extremely low temperatures (−60° to 80° C) and can be stored for years or discarded following their use as sequencing reagents. Early bacterial cloning systems only allowed for the cloning of small DNA fragments of no more then 10,000 bases (10 kb). Later attempts at cloning large DNA fragments that would facilitate the representation of entire genomes at redundant levels in single libraries were initially made using yeast as a cloning vector, but the yeast system was technically challenging, difficult to automate and produced libraries with high levels of chimeric clones.
The revolution in large fragment DNA cloning was first reported in 1990 and described a new type of single-copy plasmid vector called a Bacterial Artificial Chromosome (BAC) (Shizuya et al. 1992). The BAC system allowed for the cloning of very large pieces of DNA (100 to 300 kb) using established E. coli protocols with only moderate modification. In BAC cloning, the target substrate represents size-selected large fragment portions of partially digested DNA. The large partially digested fragments provide the ability to contiguously assemble overlapping clones into a genomic physical map. Given this level of cloning capacity, BAC libraries that represented a 10-fold redundant coverage (or more) of a large genome, like that of humans, could be developed. The first reported use of BAC libraries was for human DNA, but the technology was subsequently utilized for many animal and plant taxa.
Fig. 3. Development of a physical framework for an isolated section of a hypothetical genome. The illustration shows how overlapping large fragment clones form a contig. The addition of genetic markers to the contig is also illustrated to form the physical-genetic (genomic) framework. Entire chromosomes and genomes can be assembled via the development of these contigs which are oriented and positioned with the genetic markers.
While BAC libraries could be applied to a variety of genomic applications, their primary utility was in the development of contig-based clone maps that could be integrated with genetic maps to form an elaborate physical-genetic framework for genome sequencing (Meyers, Scalabrin, and Morgante 2004; Warren et al. 2006). In developing a contig-based clone map, the clones in a BAC library are first fingerprinted; meaning that the DNA of each clone fragment is systematically cut with one or more restriction enzymes. The fragments are then separated based on size through a process called electrophoresis. The patterns of fragmentation are then digitized and placed in a database of clone fingerprints. Clones with shared fragmentation patterns (fingerprints) are computationally assembled into sets of overlapping clones to form large reconstructed sections of chromosomes (fig. 3).
Through a process of tagging the BAC clones in a physical map with corresponding markers from a genetic map, based on sequence similarity, the physical map could be integrated with the genetic map (fig. 3). Knowledge of BAC clone and fragment size in a physical-genetic map allows for the calculation of actual physical distance or base pairs of DNA between genetic markers. This is analogous to determining the actual mileage between cities on a map. Conversely, the clone-based contigs themselves can now be positionally oriented in the genome based on the linkage-groups (corresponding to chromosomes) in the genetic map. By assembling the clone contigs into their respective linkage groups, based on their association to corresponding genetic markers, entire chromosomes can be reconstructed. The end result is a highly accurate map of the entire genome of an organism that can serve as a framework tool for a variety of applications, including the identification of genes of interest, targeted genome sequencing, and complete genome sequencing.
Sequencing Strategies Developed in the Human Genome Project
The public sector of the human genome project was a consortium of laboratories around the world located largely in the USA, England, France, and Japan. Using the physical-genetic map, the various labs were each assigned a specific set of overlapping BAC clones to sequence in a methodical clone-by-clone highly ordered strategy. Multiple locations on chromosomes were being sequenced at the same time, each initiated by a single BAC called a seed clone. Despite this technology, there are still regions of the human genome which remain unsequenced due to their highly repetitive and variable nature. These regions are so large that they cannot be bridged by a BAC clone.
Each BAC clone selected for genome sequencing became the chief substrate for DNA sequencing. This was accomplished by the physical shearing of the 100 to 300 kb BAC clone, followed by end-repair of the fragments, and cloning into a small-insert plasmid sequencing vector. The BAC sub-clones are then production sequenced en masse until about an 8- to 10-fold redundant coverage of the original BAC clone has been achieved. Following assembly of the production sequence reads, in most cases there remain gaps in the sequence that need to be closed in a process called “finishing” or “gap closure.” Gap closure often requires the use of a variety of techniques and chemistries and typically costs as much or more than the original production sequencing operation. In cases where a gap could not be closed with actual DNA sequence, it was often bridged with paired reads from both sides of the gap with a large DNA clone of known size.
This whole process of methodical genome sequencing is quite involved, time consuming, and expensive. As a result, government DNA sequencing funding strategies were changed after the human genome and several model genomes were completed.
Whole Genome Shotgun Sequencing (WGSS)
In contrast to the effort by the public sector, which did not produce a workable draft of the genome until 2001 and a near-complete final version in 2003, research scientist Craig Venter in the private sector (Celera Genomics), proposed a more rapid approach (Istrail et al. 2004; Venter et al. 2001; Weber and Myers 1997). Venter’s method employed a technique called “whole genome shotgun sequencing ” (WGSS) in which construction of an initial genetic-physical framework may be bypassed. In such a project, the entire genome is fragmented en masse and cloned as large batches of random fragments. To improve the process, multiple types of plasmid vectors and fragment sizes are cloned, providing multiple libraries for sequencing. The clones in each of the libraries are then production sequenced en masse to certain levels of genomic redundancy based on research funds. The caveat of the propaganda surrounding Venter’s “whole-genome shotgun sequencing” effort was the fact that his laboratory still relied on the use of the physical-genetic framework developed by the public sector of the human project to sort out the huge mass of random DNA sequences and sequencing contigs. This caveat, even though clearly outlined in the official journal publication (Venter et al. 2001), was never widely discussed in the popular media. Nevertheless, the concept of “whole-genome shotgun sequencing” became quite popular and was subsequently used as a cost-effective strategy for genome sequencing for a wide variety of other plant and animal genomes.
Chimpanzee Shotgun Sequence and the Human Framework
While one would think that the basic technical process of producing a genomic sequence would be free of any philosophical constraints, this is not always the case. Perhaps the most dramatic example of this is the chimpanzee genome project which consisted of an initial 5-fold redundant shotgun coverage (The Chimpanzee Genome Consortium 2005). In contrast to the human genome project, funding was limited and the project initially employed a “whole-genome shotgun sequencing” strategy that produced a 5-fold redundant coverage. However, to organize the millions of sequencing reads, the human genome physical framework was initially used as a scaffold. In other words, the chimp genomic sequence was sorted out and organized according to the human genomic framework under the assumption that chimpanzee and human are genetically similar, which evolutionists assume is due to a shared common ancestor about one to six million years ago.
One concern regarding the use of the human genome as a framework for chimpanzee is the possibility that there may be a major size discrepancy. Using flow cytometry to estimate nuclear DNA content, the human genome is widely used as a calibration standard at 7.0 picograms for a 2C diploid cell (Dolezel and Greilhuber 2010), and listed at 3.5 pg for a 1C equivalent at www.genomesize.com. At the same web site, there are five referenced estimates for chimpanzee which range from 3.46 to 3.85 for 1C; a 0 to 10 % increase in genome size compared to human. The reported average estimated genome size increase of chimpanzee over human is about 5%. Interestingly, in 2009, statistics for the chimpanzee genome sequencing effort posted on the Washington University Genome Center web site indicated that the total amount of contiguously assembled chimpanzee sequence was close to 20% more than the same parameter for the human genome. However, the sequencing statistics for chimpanzee were removed from the web in 2010 even though a new build version was announced. At the time of this writing (2011), no current chimpanzee genome assembly statistics are listed online although DNA sequence and BAC clone fingerprint data are freely available for public download.
Perhaps the most startling human-chimpanzee genome data of recent times, are the results from comparing DNA sequence from human and chimpanzee Y-chromosomes (Hughes et al. 2010). Specifically, this recent study involved the comparison of the male-specific regions of the Y chromosome (MSY). While much of the human Y chromosome has been sequenced, only the MSY region of the chimpanzee Y chromosome was sequenced to a high level of completion and then compared to the corresponding region in the human Y-chromosome.
What made this study unique was that the MSY region in chimpanzee was largely assembled and constructed based on a clone-based physical map for chimpanzee, not the human physical frame-work. This allowed for a relatively reasonable comparison of the MSY sequence between human and chimp, the first time such an apparently unbiased large-scale comparison had actually been done. The results were completely unexpected and radically contradicted the standard evolutionary dogma which pervades the scientific community. The research paper title was well chosen and a very accurate one-sentence summary of the project: “Chimpanzee and human chromosomes are remarkably divergent in structure and gene content.” Perhaps the most interesting highlight of the study was the difference in gene content. While the non-genic areas between human and chimp in the MSY region were also dramatically different, the human MSY contained 78 genes while the chimpanzee only contained 37, a 48% difference in total gene content alone. In addition, the human MSY contained 27 different classes of genes (gene families/categories) while chimpanzee contained only 18; meaning that nine entire classes or gene categories were not even present in the chimpanzee MSY region. Perhaps the best way to summarize the unprecedented project is to quote some lines from the original research report.
Here we finished sequencing of the male-specific region of the Y chromosome (MSY) in our closest living relative, the chimpanzee, achieving levels of accuracy and completion previously reached for the human MSY. By comparing the MSYs of the two species we show that they differ radically in sequence structure and gene content . . . The chimpanzee MSY contains twice as many massive palindromes as the human MSY, yet it has lost large fractions of the MSY protein-coding genes and gene families present in the last common ancestor (excerpt from abstract, Hughes et al. 2010, p. 536).
A number of autosomal comparative studies have been done using both coding and non-coding sequences. Two of the most prominent studies are worth mentioning briefly. The first is a comparative study between human chromosome 21 and chimpanzee chromosome 22; so-called homologs (Watanabe et al. 2004). The chimpanzee sequence was somewhat limited at the time, but in contrast to the recent Y-chromosome project, a physical map for chimpanzee was not utilized. Large insert clones were selected by screening libraries with human probes and only the most highly alignable human-like clones were selected. These hand selected and sequenced clones were oriented on the human physical framework with the non-alignable sections and gaps ignored. As a result, the data regarding genomic similarity was biased or constricted to those areas which were previously determined to be strong candidates for similarity.
Although the authors provide interesting data for the selected regions they analyzed, they do not commit to any definitive level of overall sequence similarity other than to say that 83% of the translated protein coding regions would produce differences in protein sequence between human and chimp. Considering that only similar DNA clones were selected, the fact that 83% of the actual coding sequence would produce different proteins is indicative of more dissimilarity than similarity. We also now know that protein translation is a complicated mix of non-protein coding DNA regulation features where a single gene under differential control can produce a wide variety of transcripts (Barash et al. 2010; Wang and Burge 2008). Nevertheless, evolutionists will cite the Watanabe et al. (2004) study as a conclusive genomic effort for high sequence similarity.
The second study of interest is a whole genome type of comparison using chimpanzee genomic sequences derived from the ends of large insert clones, called BAC-end sequences (BES) (Britten 2002). The chimpanzee sequences are first screened for anything that’s human-like and highly alignable and then the best candidates are passed along for more detailed analyses. It should also be noted that such a procedure eliminates large portions of important non-coding regulatory sequences. Sequences of selected interest are then, once again, positioned using the human physical framework and then evaluated for similarity.
The Y-chromosome project only evaluated a single isolated portion of the Y-chromosome; the only part that was readily alignable was novel in that it utilized an actual physical framework derived for the chimpanzee genome to isolate and target sequence for comparison. The section that was chosen for the Y-chromosome effort also appears to be the most readily amenable to comparative study. A physical map assembly has recently been reported for chimpanzee (Warren et al. 2006). However, the only published genomic sequence comparison between human and chimpanzee using species specific physical frameworks has been the Y-chromosome project. It would be quite valuable to evolutionists and creationists alike if un-biased large-scale autosomal comparisons between human and chimpanzee could be completed now that the resources are available. In fact, the results of the Y-chromosome study demand that similar approaches be taken for the rest of the genome.
Implications for Next Generation Sequencing Technologies
Massively parallel DNA sequencing representing next generation technologies refers to literally thousands of individual reactions conducted simultaneously by a single machine (see Mardis 2008 for a technological review). The different proprietary DNA sequencing systems being utilized are based on a single general concept; the amplification of individual DNA strands in a massively parallel (simultaneous) fashion. The strand being copied from the template fragment in each individual reaction is systematically interrogated by high precision optics such that the consecutive addition of nucleotide bases up to a threshold level is determined. In general, for each technology, the more bulk DNA sequence obtained in a single machine run (~6 to 8 hours), the shorter the individual read lengths. As mentioned previously, current systems typically produce 25 to 100 bases of high quality sequence with some companies now claiming routine reads up to 400 bases. Despite the marked reduction in read length compared to Sanger-style methodologies (still commonly used), the two primary advantages include: no DNA cloning/bacterial manipulation is required and the production of megabase quantities of DNA sequence in a single run.
The new massively parallel sequencing technology has proven ideal for the sequencing of microbial genomes, whole microbial communities (metagenomics), diverse types of transcriptomes, and eukaryotic genome re-sequencing for polymorphism detection (genetic variation). The DNA substrate for these technologies is often randomly sheared whole genome (shotgun) fragments; similar to the first step of DNA preparation used in WGSS discussed previously. Because of this, the same problems apply to the resulting genomic sequences. In fact, the problem of sorting out and aligning sequences in the genome is even worse because of the short read lengths. In other words, you will need an existing physical framework to sort out the data, particularly in eukaryotic genomes like human. While the new sequencing technologies are extremely innovative, there are caveats that must be understood to properly utilize them.
Conclusion
In the early days of biotechnology, it became apparent that humans, apes, and other mammals shared protein sequences that were very similar. In fact, many human proteins exhibit high amino acid similarity in both ape and non-primate mammalian taxa (Clamp et al. 2007). One of the primary issues of concern in various evolutionary studies is that most scientists only take into account similarities between biological sequences present in both human and apes that are pre-selected and already considered similar at some level. Also, DNA sequences that do not align well are often discarded or gaps may not be accounted for in alignment analyses. Another important consideration is whether an expressed genomic product is doing the same thing in humans as it does in apes and is it expressed in the same way? These factors are often not given proper recognition. A majority of the public and scientific community are not aware of these caveats and still told hold to the dogma that the human genome is 98 to 99% similar to chimpanzee, which is most likely not the case. The fact is that major differences between the structure of the human and a chimpanzee genomes are now being documented as the genomic resources improve.
When evaluating comparisons between genomes using DNA sequence, it is important to understand the nature of how that sequence was obtained and bioinformatically manipulated. It is not uncommon to arrange the DNA sequence of a genome for which little is known by using the genome of a hypothetical evolutionary common ancestor or ”close relative” that has better-developed genomic resources. This obviously introduces an evolutionary bias at several levels. Furthermore, sequence comparisons that have yielded similarities are typically screened DNA clones and regions selected beforehand based on some level of similarity. While many DNA sequences in eukaryotic genomes are difficult to work with due to their repetitive nature, they also contain critical regulatory features that are now appearing to be just as important as the genes themselves for proper function. Understanding the technology used to produce a genomic DNA sequence product is critical prior to making any definitive conclusions about the data in question.
Most biologists among creationists and evolutionists would expect DNA sequence similarities between humans and apes due to shared anatomical and physiological features. However, it is very likely that earlier comparative genomic studies constrained by limited resources and propelled primarily by evolutionary dogma, need to be repeated using better tools and less bias.
References
Barash, Y. et al. 2010. Deciphering the splicing code. Nature 465:53–59.
Britten, R. J. 2002. Divergence between samples of chimpanzee and human DNA sequences is 5% counting indels. Proceedings of the National Academy of Sciences of the United States of America 99 no. 21:13633–13635.
Clamp et al. 2007. Distinguishing protein-coding and noncoding genes in the human genome. Proceedings of the National Academy of Sciences of the United States of America 104, no. 40:19428–19433.
Dolezel, J. and J. Greilhuber. 2010. Nuclear genome size: Are we getting closer? Cytometry Part A 77, no. 7:635–642.
Fraser, C. M. et al. 1995. The minimal gene complement of Mycoplasma genitalium. Science 270, no. 5235:397–403.
Hobolth, A. et al. 2011. Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Research 21, no. 5:349–356.
Hughes, J. F. et al. 2010. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 463:536–539.
International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409:861–920.
International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431:931–945.
Istrail et al. 2004. Whole-genome shotgun assembly and comparison of human genome assemblies. Proceedings of the National Academy of Sciences of the United States of America 101, no. 7:1916–1921.
Kong, A. et al. 2002. A high-resolution recombination map of the human genome. Nature Genetics 31:241–247.
Mardis, E. R. 2008. Next-generation sequencing methods. Annual Review of Genomics and Human Genetics 9:387–402.
Meyers, B. C., S. Scalabrin, and M. Morgante. 2004. Mapping and sequencing genomes: Let’s get physical. Nature Reviews Genetics 5, no. 8:578–588.
Mushegian, A. R. and E. V. Koonin. 1996. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proceedings of the National Academy of Sciences of the United States of America 93, no. 19: 10268–10273.
Rogers, Y. H. and J. C. Venter. 2005. Massively parallel sequencing. Nature 437:326–327.
Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America 74, no. 12:5463–5467.
Shizuya, H. et al. 1992. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proceedings of the National Academy of Sciences of the United States of America 89, no. 18:8794–8797.
Taylor, J. 2009. Not a chimp: The hunt to find the genes that make us human. New York, New York: Oxford University Press.
The Chimpanzee Sequencing and Analysis Consortium. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87.
Venter, J. C. et al. 2001. The sequence of the human genome. Science 29, no. 5507:1304–1351.
Wang, Z. and C. B. Burge. 2008. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 14:802–813.
Warren, R. L. et al. 2006. Physical map assisted whole-genome shotgun assemblies. Genome Research 16:768–775.
Watanabe et al. 2004. DNA sequence and comparative analysis of chimpanzee chromosome 22. Nature 429:382–388.
Weber J. L. and E. W. Myers. 1997. Human whole-genome shotgun sequencing. Genome Research 7:401–409.
Wildman, D. E. et al. 2003. Implications of natural selection in shaping 99.4% nonsynonymous DNA identity between humans and chimpanzees: Enlarging genus Homo. Proceedings of the National Academy of Sciences of the United States of America 100, no. 12:7181–7188.