The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
To provide a fresh and less-biased global set of analyses, large-scale comparative DNA sequence alignments between the chimpanzee and human genomes were performed with the BLASTN algorithm. One group of experiments was conducted with query and subject low-complexity sequence masking enabled while the second set had masking parameters disabled. Each group of sub-experiments tested fifteen combinations of three different word sizes (7, 11, and 15) and five different e-values (1000, 10, 0.1, 0.001, and 0.00001) for a total of 1.2 million attempted genome-wide alignments. Individual BLASTN query jobs each involved a data set of 40,000 chimpanzee whole genome shotgun sequences (WGSS) obtained from the National Center for Biotechnology (NCBI) and queried against four different human genome assemblies (GRCH37, GRCH36, Alternate SNP Assembly, and the Celera Assembly).
The use of low complexity sequence masking had the effect of decreasing computational time about 5–6 fold, lengthening the alignments slightly, lowering the number of database hits, and lowering the percent nucleotide identity slightly. Depending on the BLASTN parameter combination, average sequence identity for the 30 separate experiments between human and chimp varied between 86 and 89%. The average chimp query sequence length was 740 bases and depending on the BLASTN parameter combination, average alignment length varied between 121 and 191 bases.
The chimp sequences were subsequently implicated by personal correspondence with NCBI staff and supporting data from this study to be pre-screened for some level of homology to the human genome. Nevertheless, excluding data for the large amount of chimp sequence that did not align, a very conservative estimate of human-chimp DNA similarity genome-wide is 86–89%. Results from this study unequivocally indicate that the human and chimpanzee genomes are at least 10–12% less identical than is commonly claimed. These results are more clearly in line with the large anatomical and behavioral differences observed between human and chimp.
Keywords: human chimpanzee similarity, genome comparison, DNA alignment, BLASTN algorithm
Introduction
A common claim that is propagated through obfuscated research publications and popular evolutionary science authors is that the DNA of chimpanzees or chimps (Pan troglodytes) and humans (Homo sapiens) is about 98–99% similar. A major problem with nearly all past human-chimp comparative DNA studies is that data often goes through several levels of pre-screening, filtering and selection before being aligned, summarized, and discussed. Non-alignable regions are typically omitted and gaps in alignments are often discarded or obfuscated.
In an upcoming paper, Tomkins and Bergman (2012) discuss most of the key human-chimp DNA similarity research papers on a case-by-case basis and show that the inclusion of discarded data (when provided) actually suggests a DNA similarity for humans and chimps not greater than 80–87% and quite possibly even less. Listed below are brief analyses of three key evolutionary human-chimp genome comparison papers that provide data which is consistent with results obtained in the present study. For a more thorough literature review on this subject, see Tomkins and Bergman (2012).
One of the first publications to compare large regions of the chimpanzee genome with human, was Britten’s lab in 2002 using an in-house Fortran computer program. The study was based on five large DNA fragments (BAC clones) from chimpanzee known to be homologous to human that were thoroughly sequenced. The total length of the DNA sequence for all 5 BACs was 846,016 bases, but only 92% of the DNA aligned to human and the paper reported on only 779,132 bases. The alignment with insertions and deletions (indels) indicated a human-chimp similarity of 95% (Britten 2002). However, when the complete sequence of all 5 BACs is included, a final DNA similarity of 87% is the final figure for the compared homologous regions between chimp and human.
In 2004, Watanabe, et al. used a variety of BAC libraries to select clones for DNA sequencing representing chimp chromosome 22. The sequence was then compared to homologous regions in human. One of the caveats, is that the chimp BACs were only chosen if they each contained 6–10 human DNA markers. These initial levels of biased pre-selection are a commonly employed technique. As is the case with a number of evolutionary publications, overall DNA alignment statistics are not given in the paper or in the supplemental information. For the aligned segments, the authors give a nucleotide substitution rate of 1.44% in, but do not provide similarity estimates to include indels. The authors indicate that there were 82,000 indels and provide a histogram showing the size distribution. Data for average indel size or total indel length was conspicuously absent. Additionally, the number of sequence gaps were given, but specific data about total gap length was absent. Despite the fact that well-sequenced orthologous regions are being compared, data that would allow the calculation of accurate DNA similarity between human and chimp is omitted. Based on estimates derived from graphical data regarding base substitutions and indels, an estimate of about 80–85% overall similarity can be inferred.
The major milestone publication regarding human-chimp genome comparison was the 2005 Nature paper from The Chimpanzee Sequencing and Analysis Consortium. Unfortunately, the comparative data were given in a highly selective and confusing format and detailed tabularized data about the alignments were absent. The majority of the paper was primarily concerned with a variety of hypothetical evolutionary analyses for various divergence rates and selective forces. However, based on the numbers given in that paper, a rough overall genome similarity between humans and chimp can be calculated. The authors state:
Best reciprocal nucleotide-level alignments of the chimpanzee and human genomes cover ~2.4 gigabases (Gb) of high-quality sequence (The Chimpanzee Sequencing and Analysis Consortium 2005, p. 71).
At this point in time, the human genome assembly was estimated to be nearly complete at 2.85 Gb and had an error rate of 1 in 100,000 bases (International Human Chimpanzee Genome Sequencing Consortium 2004). The chimp genome authors in 2005 also state:
the indel differences between the genomes thus total ~90 Mb. This difference corresponds to ~3% of both genomes and dwarfs the 1.23% difference resulting from nucleotide substitutions (The Chimpanzee Sequencing and Analysis Consortium 2005, p. 71).
By applying the indel and substitution data (4.23%) to the 2.4 gigabases of aligned human-chimp sequence, and factoring in the amount of human sequence that did not align, a maximum similarity of 80.6% can be calculated. This is a very conservative estimate because nucleotide BLAST default alignments mask large amounts of low-complexity sequence. In addition, the most recent chimp genome framework (“golden path” contiguous ENSEMBL assembly; http://uswest.ensembl.org/Pan_troglodytes/Info/Index) indicates that the chimpanzee genome is approximately 8% larger than human. The inclusion of this data would further drop the genome-wide similarity below 74% identity. For a recent review on how the chimp and human genomes were sequenced and why an understanding of these technologies is essential to interpreting DNA similarity issues, see the recent review by Tomkins (2011a).
Since the Chimpanzee Sequencing and Analysis Consortium 2005 report, comprehensive genome-wide comparisons between human and chimp have been lacking in the secular literature.
Creationist Reviews and Analyses
In general, creationist research into the area of human-chimp genome similarity has been largely limited to the interpretation of claims made in evolutionary research without fully addressing the highly selective methods used or the non-alignable data that is often omitted. Nevertheless, many important points and discoveries have been brought to light.
Prior to the completion of the chimpanzee genome project, molecular biologist David DeWitt points out that despite the supposed high DNA similarity between human and chimp, significant differences exist in cytogenetics, types and numbers of transposable elements, insertion and deletion events, gene expression patterns and mRNA splicing (DeWitt 2003). In a later report, DeWitt also demonstrates that if a 5% genome-wide difference is accepted, this level of similarity is still insufficient to support various hypothetical models for selection and common ancestry consistent with evolutionary timelines (DeWitt 2005). The rate of mutational buildup in the genome of humans was further tested in computer simulations by Sanford et al. (2008) and found to represent a serious challenge to Darwinian evolutionary timelines irrespective of reported human-chimp genome differences.
Many mutations (DNA sequence differences) separating human and chimp from a common ancestor are thought to take place in regions where the genome is non-coding, a finding recently confirmed by an evolutionary report (Polavarapu et al. 2011). While evolutionary reports of non-coding DNA differences between humans and chimps continue to emerge, the logical association between these differences and the now well-documented functional and feature-rich nature of the entire non-coding region of the human genome is dramatically down-played. The wide diversity of research into the Encyclopedia of DNA Elements (ENCODE) has spectacularly confirmed the many critical features of non-coding DNA (The ENCODE Project Consortium 2011). In the area of creationist research, biologists Woodmorappe and Batten were some of the first creationist authors to illustrate how a diversity of data in the field of non-coding DNA provided support to the genome-wide function of a wide variety of important non-coding sequence classes and DNA features (Batten 2005; Woodmorappe 2004). In a recent comprehensive review that discusses a wide variety of design features in non-coding DNA, molecular biologist and intelligent design proponent Jonathan Wells thoroughly debunks the fraudulent concept of junk DNA (Wells 2011). For a brief review on the subject associated with a summary of Wells’ book see the recent article by Tomkins (2011b).
Perhaps the greatest ongoing discrepancy between human and chimp that does not fit with the so-called high similarity claims, is the marked differences in behavior and anatomy as summarized by creationists Anderson (2007), Purdom (2006) and Wieland (2002). These obvious differences between human and chimp do not seem to correlate with the supposed claims of nearly identical DNA similarity between the taxa. In fact, a secular science writer for the BBC has recently published an entire book documenting this paradox titled Not a chimp (Taylor 2009).
While many creationist authors tentatively accepted the standard evolutionary claims regarding human chimp DNA similarity, a number of reports indicated that the “nearly identical” dogma was not as clear-cut as it seemed to be. In fact, it was indicated that evolutionary data reports on human chimp DNA similarity largely represented pre-screened data that is already know to be homologous (similar in sequence) at some level, such as highly similar protein coding sequences shared among the taxa (Tomkins 2009a, 2009b). In addition, a recent literature review combined with a bioinformatics research project, evaluated the hypothetical fusion of two chimp-like chromosomes (2a and 2b) to form human chromosome 2. This project showed that the evolutionary primate fusion paradigm was seriously flawed in a number of key respects, further discounting nearly identical DNA claims (Bergman and Tomkins 2011; Tomkins 2011c; Tomkins and Bergman 2011).
Very few large-scale bioinformatics studies comparing the human and chimp genomes exist within the creationist research community. The first report of such an analysis is briefly described by creation biologist Todd Wood in the course of a published review on human and chimp biological similarity (Wood 2006). While this report is largely a literature review, it features a brief description of Wood’s own analysis that attempts to validate the 2005 chimp genome assembly. Using deduced protein sequences from shared genes already known to be similar and, thus alignable, chimp and human are compared in large-scale amino-acid sequence alignments by Wood. Protein comparisons between electronically translated DNA coding sequences of known orthologs (genes that are similar across species) is not an accurate indicator of genome-wide DNA similarity because less than 4% of the human genome actually codes for protein (International Human Genome Sequencing Consortium 2004). More importantly, the major problem with using electronically generated proteins for comparisons is the fact that most human genes undergo alternative transcription and translation, multiple methods of exon splicing, intragene regulatory RNA coding segments, enhancer elements and many other complex transcriptional splicing code features (Barash et al. 2010; ENCODE Project Consortium 2011; Wells 2011).
More recently, Wood presented a human-chimp genome-wide comparison paper at the 2011 Creation Biology Society annual meeting and published a brief abstract of the effort (Wood 2011). Wood indicated that he used the BLASTN algorithm to align in pairwise fashion 40,000 random chimp genome sequences against the most recent version of the human genome. However, details about algorithm parameters employed or how the data was returned and evaluated were lacking. Wood apparently used standard default parameters that would have incorporated sequence masking and the compiling of multiple hit data for single query sequences—chimp sequences hitting in multiple locations in the human genome. As a result, sum totals of alignments for multiple query hits rather than comparisons between individual chimp sequence queries were reported. From the available results presented in the abstract, it appears that Wood may have opted for Megablast usage—a variant of BLASTN—that uses default parameters which includes a discontiguous word size template feature and scoring matrix. Megablast compiles the most highly similar DNA sequences— omitting a majority of the less complex and lower similarity genomic features which comprise the bulk of the human and chimp genomes. As a result, Wood’s final statistics are skewed towards extremely high identities—omitting a majority of the sequence being compared.
Regardless of whether Wood used Megablast or standard BLASTN default values, these approaches are typically only used for detecting areas of extremely high similarity and do not provide objective genome-wide alignment data. An attempt was made to repeat a smaller subset of Wood’s research using the standard default parameters for BLASTN (word size = 11, default gapping, and an e-value = 10) and only a maximum DNA identity between chimp and human of 89% was obtained (Tomkins 2011d). This value conflicts with the reported 98+% similarity given in the Wood (2011) abstract. These preliminary results reported by both Wood and Tomkins clearly show that additional research in this area using a broader range of more carefully controlled BLASTN algorithm variables is warranted.
Genome Comparison Philosophy and Approach
To perform a fresh and less biased comparison of the chimp and human genomes, a study was undertaken to query chimpanzee whole genome shotgun sequence (WGSS—same as those used by Wood 2011) against four available versions of the human genome. In theory, the chimp WGSS is supposed to be random as it is derived from physically sheared (fragmented) genomic DNA that is cloned into plasmid sequencing vectors. A compressed archive of exactly 40,000 selected WGSS reads can be downloaded from the National Center for Biotechnology (NCBI; www.ncbi.nlm.nih.gov). A ‘TRACEINFO’ xml file was included that described the chimp sequences as being fully processed— trimmed for low quality bases and contaminating vector sequence. Despite the files being listed as trace reads, they were not raw unprocessed reads as Wood claimed in his 2011 abstract. Therefore, the sequences were directly usable for querying with the Basic Local Alignment Search Tool (BLAST) algorithm (Altschul et al. 1990) without any additional processing to improve quality.
For the target database, the most recent BLAST pre-formatted human genome assembly archive files were downloaded from an FTP archive at NCBI. According to more detailed information received via personal communication with NCBI staff, the archive contains four different human genome assemblies (GRCH37, GRCH36, the Alternate Assembly and the Celera Assembly). These assemblies did not undergo any pre-masking of low-complexity sequence, thus allowing the ability to include four different complete assemblies of the entire human genome in the target database and test the effects of sequence masking.
While a majority of the genome is now known to be widely functional (Wells 2011), the usage of low-complexity sequence masking in BLASTN searches excludes many of these key genetic features. Arguments to employ low-complexity DNA sequence masking were made by the original BLAST developers (Altschul et al. 1994) based on the fact that such sequences often confound evolutionary analyses and they make the following statement regarding these DNA features:
most of these segments do not generally give meaningful alignments position by position in ways that reflect actual structural and mutational history: they evidently evolve relatively rapidly.
A lack of low-complexity sequence masking also causes the inclusion of considerable amounts of additional DNA sequence resulting in a marked increase in computational processing resources. However, improvements in computer hardware performance since 1994 make large-scale DNA analyses more feasible and the inclusion of low-complexity sequences is now readily testable. Therefore, two separate sets of experiments were employed. One set of experiments employed masking for both query and subject while the other completely disabled masking.
The heuristic BLASTN algorithm is well suited for computationally demanding searches of very large DNA databases—the goal being the local identification of regions of similarity for short segments of DNA sequence such as the individual chimp WGSS that average 740 bases each. The BLASTN algorithm works by initiating short matches based on the defined word size (number of identical DNA bases). These initial seed matches are then sequentially extended in both directions until the alignment no longer exists at a significant level (based on the preset e-value) or either of the two aligned sequences terminate. While there is abundant published literature on the usage and mechanics of the BLAST algorithm for protein-related similarity searches, minimal research exists regarding its parameter exploitation in the form of its nucleotide version for large-scale genome-wide studies. The original paper published to describe the BLAST algorithm is still one of the most informative (Altschul et al. 1990). For a more current and comprehensive review of the BLAST algorithm, see Mitrophanov and Borodovsky (2006).
To produce a comprehensive set of results, it was decided that a variety of BLASTN algorithm parameter combinations in separate computational experiments would be the most effective approach—roughly similar to those used previously by Altschul et al. (1990). Specifically, combinations of three word size parameters (7, 11, and 15) and five e-value parameters (1000, 10, 0.1, 0.001, and 0.00001) were tested. The lower the e-value that is set, the more stringent and exact the sequence match will be performed by the algorithm. Altschul et al. (1990) show in the original BLAST paper that word size and e-value are the key algorithm parameters to test in any foundational BLAST analysis.
In summary, two sets of 15 word size and e-value combinations were performed: one set of experiments employed masking for both query and subject while the other disabled it. For all 30 separate BLASTN experiments, a total of 1.2 million (40,000 queries per experiment) were made against four separate versions of the assembled human genome (~ 2.85 gigabases each).
Given that the genome-wide analyses required a large amount of cumulative and comparative data, only the top alignment for each database hit (if it existed) was returned. Gapping was disallowed for a variety of reasons. First, Altschul et al. (1990) determined that the addition of gapping strategies for alignments designed to locate regions of local similarity using BLAST was negligible. Secondly, an objective comparison among all queries negates the use of gapping with the algorithm. Finally, the top local pair-wise alignments that were obtained involved a variety of very liberal to very stringent matching parameters for word size and e-value.
For the calculation of the test-statistic that determines whether the query sequence registers a significant match score based on the pre-assigned e-value, the built-in standard nucleotide substitution matrix is used. In the NCBI version of BLASTN, this feature is not customizable. The BLASTN substitution matrix in older non-commercial WUBLAST packages has been customized previously for optimization of microarray probes (Ekland et al. 2010).
Materials and Methods
The most recent stand-alone version of the BLAST package (ncbi-blast-2.2.25+) was downloaded from the NCBI software repository (http://www.ncbi.nlm.nih.gov/guide/data-software/) and installed on a dual-quad core Intel Xeon Apple Mac G5 Desktop system with 20 gigabytes of ram. The operating system was Mac OS 10.7 with the BASH shell updated to version 4.2 for access to advanced renice capabilities for process/job control. The BLASTN jobs were semi-automated and paths and algorithm parameters set using variations of a POSIX shell script written by author Tomkins (available upon request). Output parameters of BLASTN were set to CSV (comma separated values) format for basic analyses and graphing in standard desktop spreadsheet software.
The chimp query set of 40,000 sequences was downloaded as a tar archive (chimp_traces.tar.gz) from NCBI (http://www.ncbi.nlm.nih.gov/Traces/). The chimp trace archive unpackaged as individual sequences in fasta format which were then concatenated into a single large fasta format file using standard POSIX shell commands. ‘TRACEINFO’ xml files were included that described the chimp sequences as being fully processed—trimmed for low quality bases and contaminating vector sequence. Despite the files being listed as raw trace reads, they appeared to be completely processed high-quality sequences. Therefore, the sequences were directly usable for querying with the Basic Local Alignment Search Tool (BLAST) algorithm (Altschul et al. 1990) without any additional processing to improve quality.
The most recent versions of the BLASTN preformatted human genome assembly tar archives (9 files in total; human_genomic.00 to human_genomic.08) were downloaded from the NCBI ftp site at ftp://ftp.ncbi.nih.gov/blast/db/. According to information received via personal correspondence with NCBI staff, the archives contained four different human assemblies (GRCH37, GRCH36, the Alternate Assembly and the Celera Assembly). Personal communication also verified that no pre-masking of these databases was employed. All nine archives were unpacked and deployed in a single target database directory.
The various BLASTN parameter settings that were tested and their output are shown in Tables 1 and 2. The parameter to control query sequence masking ‘-dust’ was toggled as ‘no’ or ‘yes’ (default value used = ‘20 64 1’). The target database masking parameter ‘-soft_masking’ was toggled as ‘true’ or ‘false’. Query job length varied according to masking, word size and e-value parameter settings with each job taking approximately 2 to 6 days to complete at renice settings of −10. Typically, several query jobs were run simultaneously using BLASTN CPU optimization for thread numbers (parameter ‘-num_threads’).
Results and Discussion
Maximum Identity for Human-Chimp Alignments is 86–89%
See Tables 1 and 2 for a data summary of all 30 BLASTN experiments summarizing 1.2 million attempted alignments of 40,000 chimp sequences against four different versions of the human genome.
Overall human-chimp sequence similarity for alignable regions of the two genomes varied slightly between experiment groups regarding the usage of low-complexity sequence masking. For the unmasked set of experiments, DNA similarity varied from a low of 86.4% identity to a high of 88.9%, depending on the word size and e-value parameter combination (Table 1). The usage of unmasked sequence is an important consideration given recent research suggesting that key differences between human and chimp lie within low-complexity regions of the genomes (Polavarapu et al. 2011). For the set of experiments that employed low-complexity sequence masking for both query and subject, DNA similarity varied from a low of 86.2% identity to a high of 88.8%, depending on the word size and e-value parameter combination (Table 2). The usage of masking appeared to have a slight effect on the overall sequence similarity statistics. The most noticeable difference, however, was in computational processing time which was rapidly decreased with masking enabled (data not shown).
Table 1. BLASTN results based on the complete usage of query and subject sequence (masking disabled). Data is from 40,000 WGSS chimp trace archive reads queried against four human genome assemblies (GRCh37, GRCh36, alternate assembly, and the Celera assembly). Data for the top database hit, if it existed, was returned.
| E-value Threshold | Word Size | Number of Top Hits | % Identity in Aligned Bases | Average Number Base Matches per Query Sequence | Average Number Aligned Bases per Query Sequence | Average Total Length of Query Sequence (Bases) |
|---|---|---|---|---|---|---|
| 1000 | 7 | 40,000 | 87.2 | 109 | 125 | 740 |
| 10 | 7 | 40,000 | 87.2 | 109 | 125 | 740 |
| 0.1 | 7 | 36,437 | 86.8 | 118 | 136 | 740 |
| 0.001 | 7 | 29,095 | 86.4 | 140 | 161 | 740 |
| 0.00001 | 7 | 26,108 | 86.3 | 152 | 174 | 740 |
| 1000 | 11 | 40,000 | 87.6 | 109 | 125 | 740 |
| 10 | 11 | 40,000 | 87.6 | 109 | 125 | 740 |
| 0.1 | 11 | 35,788 | 87.1 | 119 | 137 | 740 |
| 0.001 | 11 | 28,507 | 86.5 | 142 | 163 | 740 |
| 0.00001 | 11 | 25,736 | 86.4 | 153 | 176 | 740 |
| 1000 | 15 | 40,000 | 88.9 | 107 | 122 | 740 |
| 10 | 15 | 39,999 | 88.9 | 107 | 122 | 740 |
| 0.1 | 15 | 33,508 | 87.9 | 123 | 141 | 740 |
| 0.001 | 15 | 26,740 | 87.1 | 147 | 168 | 740 |
| 0.00001 | 15 | 24,392 | 86.9 | 159 | 181 | 740 |
Overall DNA sequence similarity numbers in this study fall within the range of several earlier evolutionary publications where identities of 85–87% can be calculated for omitted data (Tomkins and Bergman 2012). Based on personal communication with NCBI staff, the 40,000 chimp sequences were considered to most likely be pre-screened and already known to be homologous to humans at some level, although this could not be verified by additional inquiries. Given the fact that under several algorithm parameter combinations (Tables 1 and 2), all 40,000 sequences had positive hits on the human genome(s), it is highly likely that the chimp query sequences were pre-screened for homology to human DNA. In addition, a large amount of data outside the aligned areas of each WGSS chimp sequence was omitted by the algorithm. Therefore, a maximum identity of about 86–89% is an extremely conservative and fair estimate. These data spectacularly confirm that on a whole-genome basis, the often-touted estimates of 98–99% similarity between humans and chimps are completely inaccurate.
Table 2. BLASTN results based on the usage of low-complexity sequence masking for both query and subject. Data is from 40,000 WGSS chimp trace archive reads queried against four human genome assemblies (GRCh37, GRCh36, alternate assembly, and the Celera assembly). Data for the top database hit, if it existed, was returned.
| E-value Threshold | Word Size | Number of Top Hits | % Identity in Aligned Bases | Average Number Base Matches per Query Sequence | Average Number Aligned Bases per Query Sequence | Average Total Length of Query Sequence (Bases) |
|---|---|---|---|---|---|---|
| 1000 | 7 | 40000 | 87.1 | 109 | 125 | 740 |
| 10 | 7 | 40000 | 87.1 | 109 | 125 | 740 |
| 0.1 | 7 | 36111 | 86.7 | 119 | 136 | 740 |
| 0.001 | 7 | 28294 | 86.2 | 143 | 164 | 740 |
| 0.00001 | 7 | 25280 | 86.2 | 155 | 178 | 740 |
| 1000 | 11 | 39999 | 87.5 | 108 | 124 | 740 |
| 10 | 11 | 39997 | 87.5 | 108 | 124 | 740 |
| 0.1 | 11 | 34808 | 86.9 | 121 | 139 | 740 |
| 0.001 | 11 | 26901 | 86.2 | 148 | 169 | 740 |
| 0.00001 | 11 | 24264 | 86.2 | 159 | 183 | 740 |
| 1000 | 15 | 39997 | 88.8 | 106 | 121 | 740 |
| 10 | 15 | 39985 | 88.8 | 106 | 121 | 740 |
| 0.1 | 15 | 31361 | 87.5 | 129 | 147 | 740 |
| 0.001 | 15 | 24349 | 86.6 | 158 | 180 | 740 |
| 0.00001 | 15 | 22583 | 86.6 | 167 | 191 | 740 |
Effects of Word Size and E-value
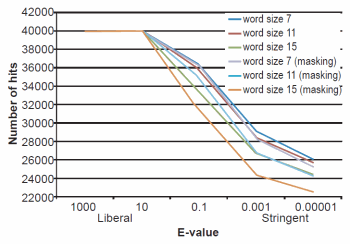
Fig. 1. BLASTN results depicting the relationship between e-value and number of hits obtained. Maximum number of hits that could obtained were 40,000.
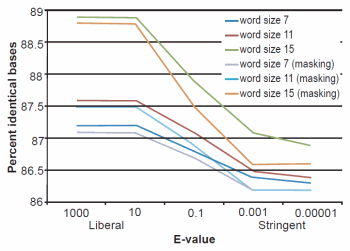
Fig. 2. BLASTN results depicting the relationship between e-value and average percent sequence identity.
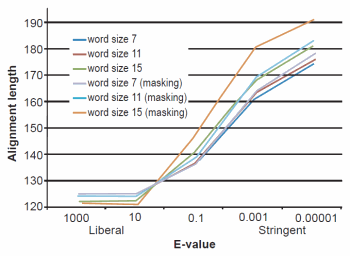
Fig. 3. BLASTN results depicting the relationship between e-value and average alignment length.
The effect of word size was rather marked and the algorithm trends were in strong agreement with those described previously by Altschul et al. (1990). See Figs. 1, 2, and 3 for a graphical depiction showing the effects of word size, e-value, and sequence masking across all experiments.
Across all word sizes, there was clearly a general trend of computational trade-offs. As e-value became more stringent, less hits were achieved (Table 1), the percent average identity of the alignments was less (fig. 2), and the average alignment length increased (fig. 3). These effects, however, did not occur until the e-value dropped below 10. For all practical purposes, the 10 and 1000 e-values produced the same results across word sizes (Tables 1 and 2; figs. 1, 2, and 3). As mentioned previously, virtually all 40,000 sequences produced hits using liberal matching parameters indicating that the query sequences were previously screened for homology to human, an observation that was largely confirmed by information provided through email correspondence with NCBI staff. Of course, a significant trade-off is that the alignments produced with liberal matching parameters, were also much shorter in length. The usage of higher levels of stringency lengthened the alignments considerably, but also lowered the percent identity and the number of positive hits on the database.
Perhaps one of the most interesting aspects of the BLASTN query experiments was the fact that even under the conditions which produced the longest alignments, only 24% (181 bases—no masking) and 26% (191 bases—masking) on average were obtained (out of 740 bases). The most liberal parameters which produced the highest sequence identities and greatest number of hits had only 16% of the 740 bases aligning.
Default BLASTN Parameter Results
The standard recommended default parameters for BLASTN listed in the help material on the NCBI web site target several search conditions. For standard nucleotide BLAST, a default word size of 11 with an e-value of 10 is used in combination with sequence masking. The default parameters in this study (Table 1) produced a full 40,000 hits and was essentially the same as using an e-value of 1000. At these settings, average sequence identity was 87.6% for the aligned regions of each hit. Average alignment length, however, was at the short end of the spectrum at 125 out of 740 bases.
NCBI help material also recommends a word size of 7 with an e-value of 1000 (and no masking) for short or near-exact matches, typically needed for specific applications where precise target sequence is required to develop primers for PCR-based lab studies. In general, these parameters facilitated the alignment of all 40,000 sequences, produced identities of 87.2% and short alignments of 125 bases.
It should be noted that both of the above default parameter recommendations by NCBI are designed to facilitate the usage and speed of on-line searches at the NCBI web tool BLAST server (www.ncbi.nlm.nih.gov/BLAST). Links to various help pages can also be accessed via the online BLAST server site.
In regards to the broad range of BLASTN experiments conducted in this study and the type of query application that they were applied to, there is limited published information available. Clearly, for future studies of this type, a comprehensive range of results can be achieved by utilizing a constant word size of about 15 in combination with e-values of 10 to 0.00001, thus reducing the number of experiments and computational resources involved.
BLAST Software Options for Genome-Wide Queries
The present study used 40,000 WGSS chimp sequences of about 740 bases on average that were queried against a database consisting of four different variants of the human genome assembly. Clearly this was a computationally intense effort that could not be performed on the NCBI BLAST web server given it’s restrictions on job size and algorithm parameter manipulation. In addition, the use of the BLAT alignment tool (BLAST-like alignment tool; Kent 2002), as employed by the UCSC Genome Browser (http://genome.ucsc.edu/), would have also been unsuitable for several reasons. First, the BLAT algorithm uses an indexed database that has low-complexity sequence omitted. Because BLAT does not directly compare sequence against sequence by using an indexing system and only returns highly identical hits, the current author did not seek to install it locally as a web-server and employ it for the present study. In fact, the UCSC web site makes the following statement regarding BLAT limitations on it’s “About BLAT” section of the BLAT server page (http://genome.ucsc.edu/cgi-bin/hgBlat?command=start).
BLAT on DNA is designed to quickly find sequences of 95% and greater similarity of length 25 bases or more. It may miss more divergent or shorter sequence alignments. It will find perfect sequence matches of 25 bases, and sometimes find them down to 20 bases.
Finally, the ability to fully exploit the many parameters available with the BLASTN algorithm and to query actual DNA sequence against DNA sequence is best accomplished with local command-line usage of the NCBI BLAST suite.
Summary and Conclusion
Large-scale comparative DNA sequence analyses were conducted between the chimp and human genomes using the BLASTN algorithm in 30 separate experiments. The individual experiments involved the use of different e-value and word size combinations under both low-complexity sequence masking and non-masking conditions for a total of 1.2 million attempted alignments. In addition to the testing of sequence masking, fifteen combinations of three different word sizes (7, 11, and 15) and five different e-values (1000, 10, 0.1, 0.001, and 0.00001) were evaluated. The top alignment hits and their associated values in each experiment were returned in each experiment.
The query data comprised a set of 40,000 chimp whole genome shotgun sequences (WGSS) obtained from the National Center for Biotechnology (NCBI) that were subsequently implicated by personal correspondence with NCBI staff and supporting data from this study to be pre-screened for homology to the human genome. The chimp sequences were queried in 30 separate experiments against four different high-quality human genome assemblies (GRCH37, GRCH36, Alternate SNP Assembly, and the Celera Assembly).
The use of low complexity sequence masking had the effect of decreasing computational time about 5 to 6 fold, lengthening the alignments slightly (0 to 12 bases), lowering the number of database hits (0 to 2,391 hits), and lowering the percent nucleotide identity slightly (0.1–0.5%).
Depending on the BLASTN parameter combination, average sequence identity for the thirty separate experiments between human and chimp varied between 86 and 89%. The average chimp query sequence length was 740 bases and depending on the BLASTN parameter combination, average alignment length varied between 121 and 191 bases.
Excluding data for the number of clones that did not align or the large amount of bases within clones that did not align, an unbiased conservative estimate of genome-wide human-chimp DNA similarity is not more than 86–89% identical. The conservative nature of these estimates is further noted by the fact that the 40,000 sequence chimp sequences that were tested, represent pre-selected homologous sequence already known to align to the human genome.
References
Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. Journal of Molecular Biology 215:403–410.
Altschul, S. F., M. S. Boguski, W. Gish, and J. Wootton. 1994. Issues in searching molecular sequence databases. Nature Genetics 6:119–129.
Anderson, D. 2007. Decoding the dogma of DNA similarity. Retrieved from http://creation.com/decoding-the-dogma-of-dna-similarity.
Barash Y., J. A. Calarco, W. Gao, Q. Pan, X. Wang, O. Shai, B. J. Blencowe, and B. J. Frey. 2010. Deciphering the splicing code. Nature 465:53–59.
Batten, D. 2005. No joy for junkies. Journal of Creation 19, no. 1:3.
Bergman, J. and J. Tomkins. 2011. The chromosome 2 fusion model of human evolution—part 1: Re-evaluating the evidence. Journal of Creation 25, no. 2:106–110.
Britten, R. J. 2002. Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proceedings of the National Academy of Sciences of the United States of America 99, no. 21:13,633–13,635.
DeWitt, D. A. 2003. Greater Than 98% Chimp/Human DNA Similarity? Not Any More. TJ 17, no. 1:8–10.
DeWitt, D. A. 2005. Chimp Genome Sequence Very Different from Man. TJ 19, no. 3:4–5.
Eklund, A. C., P. Friis, R. Wernersson, and Z. Szallasi. 2010. Optimization of the BLASTN substitution matrix for prediction of non-specific DNA microarray hybridization. Nucleic Acids Research 38:e27. doi: 10.1093/nar/gkp1116.
International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431:931–945.
Kent, J. 2002. BLAT—The BLAST-like alignment tool. Genome Research 12, no. 4: 656–664.
Mitrophanov, A. Y. and M. Borodovsky. 2006. Statistical significance in biological sequence analysis. Briefings in Bioinformatics 7, no. 1:2–24.
Polavarapu, N., G. Arora, V. K. Mittal, and J. F. McDonald. 2011. Characterization and potential functional significance of human-chimpanzee large INDEL variation. Mobile DNA 2:13 doi:10.1186/1759-8753-2-13.
Purdom, G. 2006. If human and chimp DNA are so similar, why are there so many physical and mental differences between them? Answers 1, no. 2:64–65. Retrieved from http://www.answersingenesis.org/articles/am/v1/n2/human-and-chimp-dna.
Sanford, J., J. Baumgardner, W. Brewer, P. Gibson, and W. Remine. 2008. Using numerical simulation to test the validity of neo-Darwinian theory. In Proceedings of the Sixth International Conference on Creationism, ed. A. A. Snelling, pp. 165–175. Pittsburgh, Pennsylvania: Creation Science Fellowship, and Dallas, Texas: Institute for Creation Research.
Taylor, J. 2009. Not a chimp: The hunt to find the genes that make us human. New York, New York: Oxford University Press.
The Chimpanzee Sequencing and Analysis Consortium. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87.
The ENCODE Project Consortium. 2011. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLOS Biology 9, no. 4: e1001046. doi:10.1371/ journal.pbio.1001046
Tomkins, J. 2009a. Human-chimp similarities: Common ancestry or flawed research? Acts & Facts 38, no. 6:12.
Tomkins, J. 2009b. Common DNA sequences: Evidence of evolution or efficient design? Acts & Facts 38, no. 8:12–13.
Tomkins, J. 2011a. How genomes are sequenced and why it matters: Implications for studies in comparative genomics of humans and chimpanzees. Answers Research Journal 4:81–88. Retrieved from www.answersingenesis.org/articles/arj/v4/n1/implications-for-comparative-genomics.
Tomkins, J. 2011b. The junk DNA myth takes a well-deserved hit. Journal of Creation 25, no. 3:23–26.
Tomkins, J. 2011c. Ongoing telomere research at odds with human-chimp chromosome 2 model. Acts & Facts 40, no. 11:6.
Tomkins, J. 2011d. Evaluating the human-chimp DNA myth— new research data. Acts & Facts 40. no. 10:6.
Tomkins, J., and J. Bergman. 2011. The chromosome 2 fusion model of human evolution—part 2: Re-analysis of the genomic data. Journal of Creation 25, no. 2:111–117.
Tomkins, J., and J. Bergman. 2012. Genomic monkey business—estimates of nearly identical human-chimp DNA similarity revaluated using omitted data. Journal of Creation (in press).
Watanabe, A. F. et al. 2004. DNA sequence and comparative analysis of chimpanzee chromosome 22. Nature 429: 382–388.
Wells, J. 2011. The myth of junk DNA. Seattle, Washington: Discovery Institute Press.
Wieland, C. 2002. Furry little humans? Creation 24, no. 3:10–12.
Wood, T. C. 2006. The chimpanzee genome and the problem of biological similarity. Occasional Papers of the BSG, No. 7, pp. 1–18.
Wood, T. C. 2011. The chimpanzee genome is nearly identical to the human genome. Creation Biology Society Annual Conference Abstracts 2011, vol. 1: 24-25. (http://www.bryancore.org/jcts/index.php/jctsb/article/view/8)
Woodmorappe, J. 2004. Junk DNA indicted. Creation Ex Nihilo Technical Journal 1, no. 81:27–33.