The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
Because bacteria, archaea, and eukarya contain unique mosaics of genetic features and biochemical similarities, it has been notoriously difficult for evolutionists to infer the molecular biological properties of a first or last eukaryotic common ancestor. Eukarya share similarities to both domains of prokaryotes (Bacteria and Archaea) while also exhibiting many innovative molecular features found in neither. Nevertheless, evolutionists postulate that some sort of mythical bacterial-archaeal precursor gave rise to the first eukaryotic cell. In a previous report, we showed that a vast chasm exists between archaea and eukarya in regard to basic molecular machines involved in DNA replication, RNA transcription, and protein translation. The differences in information processing mechanisms and systems are even greater between bacteria and eukarya, which we elaborate upon in this report. Based on differences in lineage-specific essential gene sets and in the vital molecular machines between bacteria and eukarya, we continue to demonstrate that the same unbridgeable evolutionary chasms exist—further invalidating the myth of eukaryogenesis.
Keywords: Origin of species, origin of eukarya, eukaryogenesis, molecular biology, molecular evolution, tree of life, First Eukaryotic Common Ancestor, FECA, Last Eukaryote Common Ancestor, LECA
Disclaimer: Regarding author C. Tan, the opinions expressed in this article are the author’s own and not necessarily those of the University of Missouri.
Introduction
What is life? Where did life come from? Are all life forms phylogenetically linked via the great “Tree of Life . . . with its ever branching and beautiful ramifications” (Darwin 1859; NSF 2010)? Can we reconstruct the history of life according to our knowledge of extant species?
Mountains of computer-generated phylogenetic trees and massive federal funding makes it appear that defining the “Tree of Life” (TOL) is within reach. In order to build the Tree of Life, the National Science Foundation (NSF) has awarded 275 projects since 2002, with $177,738,326.00, first under the name “Assembling the Tree of Life (ATOL)” from 2002 to 2013 and then under the “Genealogy of Life (GoLife)” from 2014 (Supplemental Table). In the past couple of years, several computer programs have allowed for visualizing the TOL, branches and leaves, on computers or cell phones (Kumar and Hedges 2011; Page 2012; Rosindell and Harmon 2012). These trees (http://www.onezoom.org and http://www.timetree.org), completed with time of branching and number of species at each node, are extremely impressive. Does this mean that we have solved the mystery of life, including the origin of eukarya, that is, eukaryogenesis, which is a big puzzle in the history of life and which various evolutionary models have been proposed to explain? Several reviews from both secular and creationist authors have recently been published discussing this whole state of affairs (Rochette, Brochier-Armanet, and Gouy 2014; Tomkins and Bergman 2013).
Surprisingly, a casual scan of the scientific literature studying the molecular phylogeny of life will show that one can draw not only one great tree but many, in fact, embarrassingly too many (Koonin, Wolf, and Puigbo 2009; Koonin, Puigbo, and Wolf 2011; Puigbo, Wolf, and Koonin 2009, 2012, 2013). Different molecules generate different trees, and even different regions of the same molecule can generate different trees. “As the sequences from genome projects accumulate, molecular data sets become massive and messy, with the majority of gene alignments presenting odd (patchy) taxonomic distributions and conflicting evolutionary histories . . . the expected proportion of genes with genuinely discordant evolutionary histories has increased from limited to substantial . . . If phylogenomic analysis is the objective, these discordant markers are usually removed from the data set in order to improve resolution of the tree” (Leigh et al. 2011, pp. 572, 577).
More and more studies have demonstrated that even the most phylogenetically favorable biological sequences refuse to fit into one single tree; they typically end up a forest, although often they are not presented that way (Ebersberger et al. 2007; Puigbo, Wolf, and Koonin 2009; Rochette, Brochier-Armanet, and Gouy 2014). For example, early last year, Rochette and colleagues reported a thorough phylogenomic analysis of eukaryotic proteins that have prokaryotic homologs (Rochette, Brochier-Armanet, and Gouy 2014). They identified 475 such protein families in which eukarya are monophyletic from 234,892 protein families from 820 bacteria, 62 archaea, and 64 eukarya. Since 65 of these 475 families contain two to four Last Eukaryote Common Ancestor (LECA) clades, defined as clusters of homologs composed of proteins from bacteria and/or archaea and two groups of eukarya, a total of 554 LECA clades were obtained. To reduce the size of the trees and the taxonomic biases, they selected 144 bacterial and 39 archaeal genomes. For each LECA clade, ten eukaryotic organisms were selected. The authors then reconstructed the phylogenetic trees and found that 434 (78%) of the 554 LECA clades remained monophyletic for eukarya with more than 50% nonparametric bootstrap support (Fig. 1A). Therefore, 311 (~42%) out of the 744 phylogenetic trees generated show that eukaryotes are not monophyletic (191 in the initial phylogenetic construction, 120 in the reconstruction with the reduced data set). Based on extended topological criteria called configurations, Rochette and colleagues concluded that 56% (243) of the 434 (probably 433, see note in the legend of Fig. 1) LECA clades have bacterial origin, 28% (121) archaeal origin, 0.7% (3) “three-domain” configuration (Archaea, Bacteria, and Eukarya all monophyletic), and 15% (67) have tangled phylogeny (Archaea and Bacteria appeared mixed) (Rochette, Brochier-Armanet, and Gouy 2014) (Fig. 1B). Furthermore, of the 243 LECA clades with bacterial origin, only 44 (18%) could be traced to a specific bacteria phylum (41 to Alphaproteobacteria, two to Cyanobacteria, and one to Verrucomicrobiae) (Fig. 1C). Note that the authors found that the “trees were extremely heterogeneous in terms of species content, number of paralogs per genome, branching patterns, as well as in terms of branch length and bootstrap support distributions among branches.” In fact, no two trees were the same. This echoes the conclusion of an earlier study by Puigbo and colleagues, who found that no two trees in their 102 nearly universal trees were the same (Puigbo, Wolf, and Koonin 2009). Thus, even with the most extreme data filtering, no single consensus tree could be obtained in either of these studies.
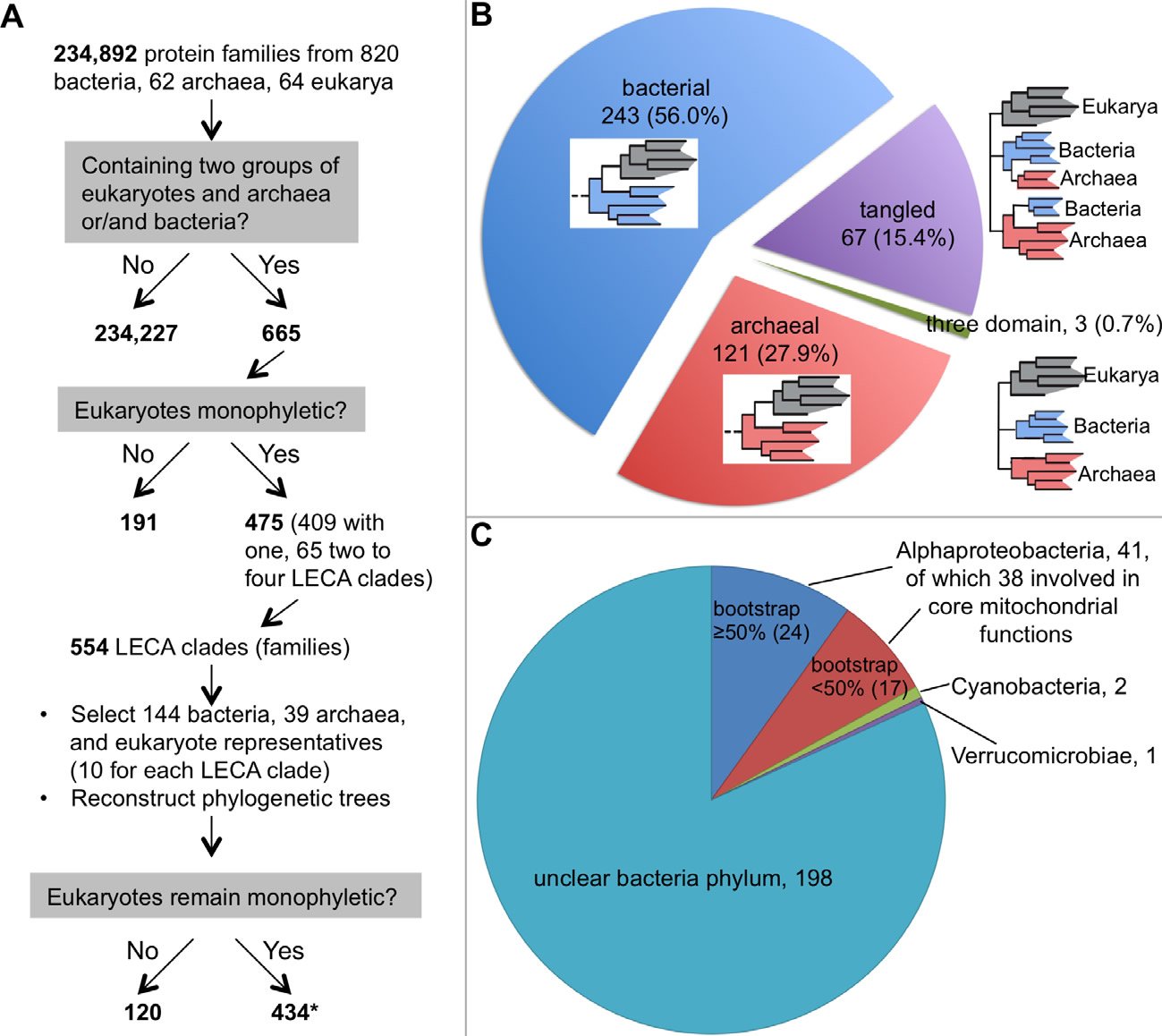
Fig. 1. A phylogenetic study of the origin of eukaryotes. A. A flow chart showing how Rochette and colleagues chose genes for their phylogenetic analysis. Note that data selection, including the removal of genes that do not have phylogenetic signals or genes that are supposed to be laterally-transferred, is a common practice among evolutionists. B. The distribution of the 434 phylogenetic trees in which eukaryotes are monophyletic. A schematic phylogenetic configuration is either included within the correspondent pie of each group or next to it. Eukaryotes are shaded gray, bacteria blue, and archaea red. C. The distribution of the phylogenetic trees with eukaryotes having a bacterial origin. Although this group is supposed to have 243 trees, we could find information for only 242 of them in the original article about their belonging to bacterial phyla (Rochette, Brochier-Armanet, and Gouy 2014).
*:This is probably 433 because the total phylogenetic trees constructed equal to 744 (665+554−475), which should include trees showing eukaryotes not monophyletic (311) and those monophyletic (744−311=433). This one tree difference may account for the tree that has eukaryotes with bacterial origin but no information could be found.
Why so many different trees, even after carefully discarding all the molecules (~42%) that make eukarya appear polyphyletic, or non-phyletic (Fig. 1A)?
In this article, we seek to demonstrate that an essential reason for the resistance of all molecules to fit onto one tree is that no such tree accurately depicts the history of life. More specifically, we show that eukarya could not have evolved from prokaryotes. We base our argument on two observations: distinct differences in lineage-specific essential gene sets and a comparison of the vital molecular machines involved in DNA replication, transcription, and translation between bacteria and eukarya. In a companion paper published prior to this report, we took a similar approach to a comparison of the information processing machinery between archaea and eukarya (Tan and Tomkins 2015).
Lineage-specific Essential Genes
Genes that are required for the viability of an organism are called essential genes. An organism dies when any one of its essential genes does not function properly. Many studies have shown that it takes very little effort to find a lethal mutation, a change in the DNA that makes an organism inviable. For example, seven lethal point mutations have been identified in the tiny Drosophila gene flapwing (flw), which is one of the estimated 14,600 genes in fruit fly Drosophila melanogaster, and this gene occupies less than 0.02 percent of the Drosophila genome (Yamamoto et al. 2013).
Hundreds of genes are necessary to sustain even the simplest life forms. For example, the organism with the smallest known genome that can constitute a cell, the pathogenic bacterium Mycoplasma genitalium (M. genitalium), contains 381 essential genes, 79% of its annotated 482 protein-coding genes (Glass et al. 2006) (Table 1, Fig. 2A). Much like the case of the flw lethal mutations with respect to the survival of Drosophila, lack of function in any one of these 381 genes will render M. genitalium inviable even “in an environment that is free from stress and provides all necessary nutrients” (Glass et al. 2006).
| organism | M. genitalium | E. coli K-12 | S. cerevisiae | Mus musculus | |
| protein genes in the genomea | 482 | 4800 | 5770 | ~23,000 | |
| total essential genesb | 381 | 712 | 1110 | >2618 | |
| analyzed genesc | 475 | 3527 | 5635 | 6038 | |
| analyzed essential genesc | 381 | 604 | 1049 | 2618 | |
| apparent age of analyzed essential genesd | |||||
| cellular organisms | 91 | 121 | |||
| Bacteria | 90 | 124 | |||
| Mollicutes | 47 | ||||
| Proteobacteria | 61 | ||||
| Gammaproteobacteria | 55 | ||||
| Enterobacteriaceae | 182 | ||||
| Eukaryota | 505 | 856 | |||
| Fungi/Metazoa group | 96 | 248 | |||
| Fungi | 78 | ||||
| Ascomycota | 67 | ||||
| Saccharomycetes | 110 | ||||
| Metazoa | 996 | ||||
| Chordata | 330 | ||||
| Mammalia | 54 | ||||
| Not assigned | 153 | 61eg | 97fg | 61 | |
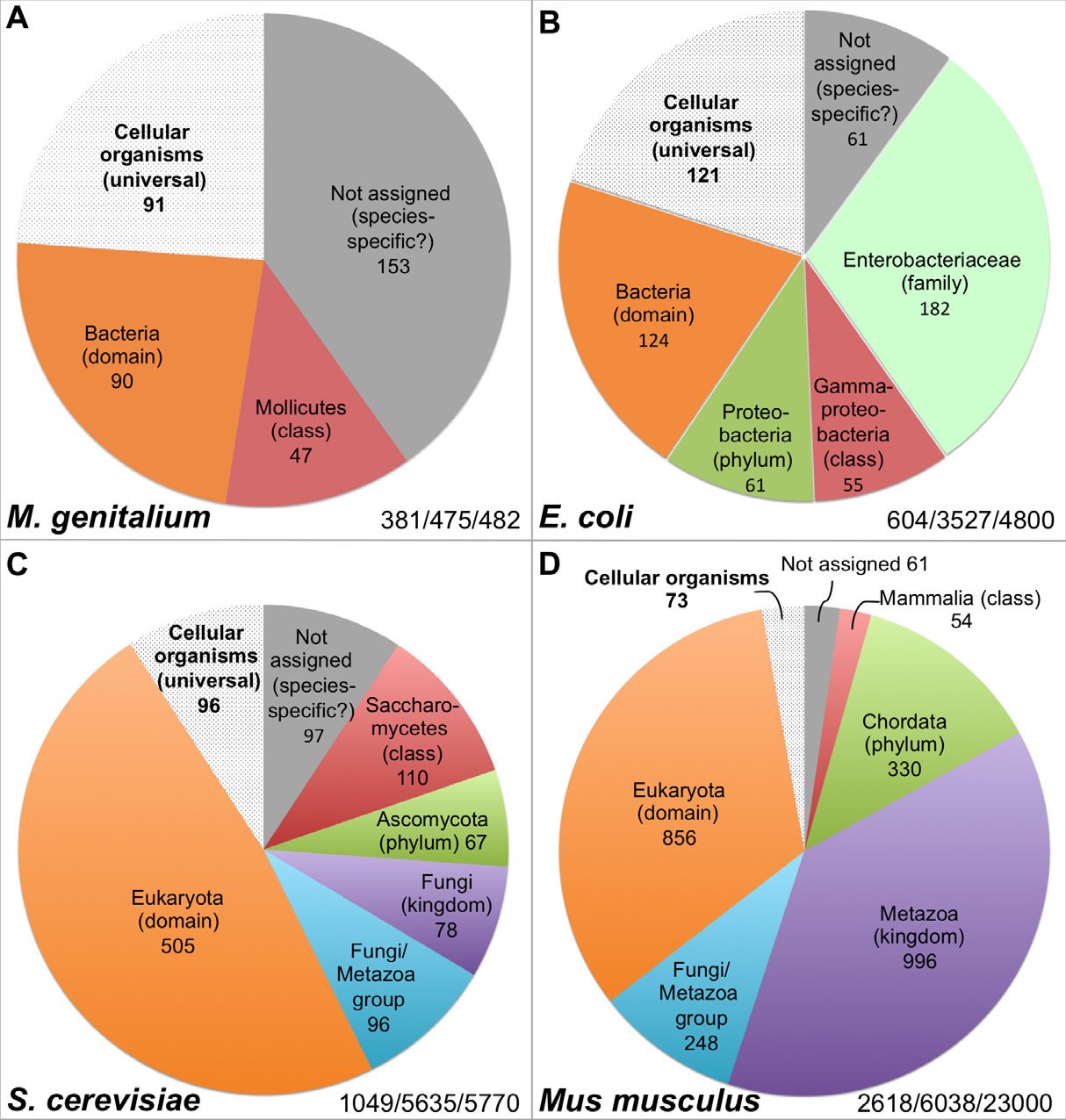
Fig. 2. Distribution of the essential genes of four organisms. The numbers at the right bottom in each panel represent the essential genes identified/genes analyzed in the study/total number of protein coding genes encoded in the genome. All data are from the online gene essentiality database (OGEE, http://ogeedb.embl.de), which is composed of large scale/genome wide analyses. The distributions of the various groups were obtained by running analyses of specific data sets with the feature “phyletic age” on the OGEE website. A: M. genitalium. OGEE dataset 357. B: E. coli. OGEE dataset 367. C: S. cerevisiae. OGEE dataset 350. D: Mus musculus. OGEE dataset 349. Definition of essentiality for both M. genitalium and E. coli: genes whose mutants cannot be obtained from the mutagenesis library, for S. cerevisiae: genes whose removal result in a lethal phenotype (growth inhibition), for Mus musculus: genes whose removal result in a lethal or infertile phenotype.
It is worth mentioning that there are many genes that, when individually deleted, would not kill an organism, but result in the death of the organism when deleted along with another nonessential gene. This well-known genetic phenomenon is called synthetic lethality (Tucker and Fields 2003). Therefore, we do not know how many additional genes in the M. genitalium genome are required for its survival, once synthetic lethality is considered. In addition, all organisms tested for gene essentiality are done in artificial and carefully controlled environments that are not indicative of natural conditions.
Current data suggest that many essential genes are lineage-specific. For example, the bacterium Escherichia coli (E. coli) contains 4800 genes, of which 712 are known to be essential. Thus, the number of essential genes of E. coli (712) is larger than the total number of genes encoded by the M. genitalium genome (482) and not all essential genes of M. genitalium have E. coli counterparts, although both organisms belong to the same bacterial domain (Glass et al., 2006). For instance, 47 of the M. genitalium essential genes have homologs only in Mollicutes, a bacterial class that E. coli does not belong to (Fig. 2A, Table 1). Interestingly, like the essential genes of M. genitalium, the majority of the E. coli essential genes do not have any eukaryotic homologs and some of them actually only have homologs in the Enterobacteriaceae family which M. genitalium does not belong to (Fig. 2A and B, Table 1), and a few are limited to E. coli species. In fact, only a small portion of the essential genes have homologs across the three domains of life, or belong to the group of cellular organism genes based on their inferred phyletic age or the hypothesized evolutionary origin of a gene, which is typically defined by the evolutionarily most distant species where homologous biological sequences can be found (Chen et al. 2012; Wolf et al. 2009). It appears that the more complicated an organism is, the smaller that portion of universal essential genes becomes, from the 22.3% of E. coli essential genes, to 9.2% of yeast, to 2.8% of mouse (Fig. 2 B–D). Thus, the vast majority of the yeast Saccharomyces cerevisiae (S. cerevisiae) essential genes are eukaryotic-specific; they do not have bacterial homologs (Fig. 2C, Table 1). As the case with E. coli, some of the yeast essential genes are limited to a specific phylum or class. Similarly, mouse (Mus musculus) essential genes are mostly eukaryotic-specific, many are specific to the animal kingdom, and some have homologs only in mammals (Fig. 2D, Table 1).
We still do not know how many of our ~30,000 human protein-coding genes are essential (Wijaya et al. 2013). So far, 118 have been identified as essential (Liao and Zhang 2008), but this number is undoubtedly far below the actual number. We cannot do lab experiments on humans with essential genes like we can with mice and we cannot predict whether a human gene is essential or not based on the essentiality of its mouse orthologs because they often lead to different phenotypes (Liao and Zhang 2008). Wilcox and colleagues found that 31% of implanted fetuses died during pregnancy (Wilcox et al. 1988), suggesting that many human genes must be intact to generate a viable human baby.
Note that the exact number belonging to different groups of genes may change with the discovery of more genes in currently uncharacterized organisms. However, this will not alter the conclusion that most bacterial essential genes do not have eukaryotic homologs, that the vast majority of the eukaryotic essential genes are unique to the eukaryotic domain, and that some essential genes are restricted to specific phylum or order or even species.
From the viewpoint of evolutionists, it is puzzling that so many essential genes have a limited distribution. As mentioned earlier, an organism cannot survive unless all its essential genes are functional. Therefore, it is impossible for organism “A” to evolve into another organism, “B,” unless all of B’s essential genes have homologs in A. Therefore, based on the fact that most of the eukaryotic essential genes do not have a bacterial homolog, we argue that eukarya could not have evolved from bacteria.
Is it really possible for an essential gene to duplicate and for the duplicate to evolve into a different essential gene that no longer looks much like the first and the first to be later lost, thus enabling organisms A and B to have the same essential function covered by two seemingly different genes? A thorough study of mouse duplicate genes by Liao and Zhang (2007) suggests this scenario is unlikely. Liao and Zhang “analyzed nearly 3900 individually knocked out mouse genes and discovered that the proportion of essential genes is approximately 55% in both singletons and duplicates” (Liao and Zhang 2007, p. 378) and suggested “that mammalian duplicates rarely compensate for each other, and that the absence of phenotypes in mice deficient for a duplicate gene should not be automatically attributed to paralogous compensation” (Liao and Zhang 2007, p. 378). A similar phenomenon has been observed in the studies of yeast ribosomal proteins; the paralogous ribosomal proteins are functionally distinct, even though some of them are nearly identical in protein sequence (Komili et al. 2007).
Information Processing Molecular Machines
As Bruce Alberts said, the cell is “a factory that contains an elaborate network of interlocking assembly lines, each of which is composed of a set of large protein machines” (Alberts 1998, p. 291). Not only do all cells contain sophisticated nano-machines, but different organisms may use distinct mechanisms or molecules to accomplish the same tasks. The finding of non-homologous parts being used to fulfill homologous functions is counter to what one would expect from the universal common descent theory.
Before we plunge into the details of any molecular machines, we would like to mention a few well-known facts about our example organisms bacteria E. coli, a representative of prokaryotes, and yeast S. cerevisiae, a representative of eukarya. We chose E. coli and S. cerevisiae not because anybody claimed that S. cerevisiae or any other eukaryote evolved from E. coli but because the same conclusion can be reached with any other pair of prokaryote and eukaryote and because E. coli and S. cerevisiae provide the advantage of being the most studied organisms.
Like most prokaryotes, E. coli has a circular chromosome. Because it lacks a nucleus, its DNA duplication, transcription, and translation all occur in the same compartment. In contrast, S. cerevisiae, like any other eukaryote, has a membrane-bound nuclear compartment (called a nucleus) that houses its chromosomes. Like all other eukarya, S. cerevisiae has its DNA organized into multiple linear chromosomes (16 pairs). It has a total genome size of 12 million bases or 12 Mb, more than two times the 4.6 Mb of the E. coli genome. S. cerevisiae is also one of the major eukaryotic model organisms for understanding cellular and molecular processes. In eukarya, DNA replication and transcription occurs in the nucleus, while translation occurs in the cytosol, also called the cytoplasm. Both E. coli and S. cerevisiae replicate DNA, transcribe DNA into RNA, and translate RNA into protein, yet the molecular nanomachines they use for replication, transcription, and translation are not at all interchangeable.
A. DNA replication machines used by cells to duplicate their genomic DNA
DNA replication can be divided into three steps: initiation, elongation, and termination (O’Donnell, Langston, and Stillman 2013). During initiation, origin-binding proteins bind the initiation site of replication (normally known as the origin of replication or the replication origin), which are specific chromosomal locations at which DNA replication is initiated, and then a small A/T-rich region of the origin will be unwound, or melted. Helicases, which are responsible for separating the double-stranded DNA during DNA replication, are loaded on the DNA by helicase loaders. Finally, primases, which are RNA polymerases, are recruited to synthesize a short RNA primer because DNA polymerase cannot initiate DNA synthesis without a primer (Fig. 3A). During elongation, DNA polymerases will take over the job started by the primase and synthesize DNA.
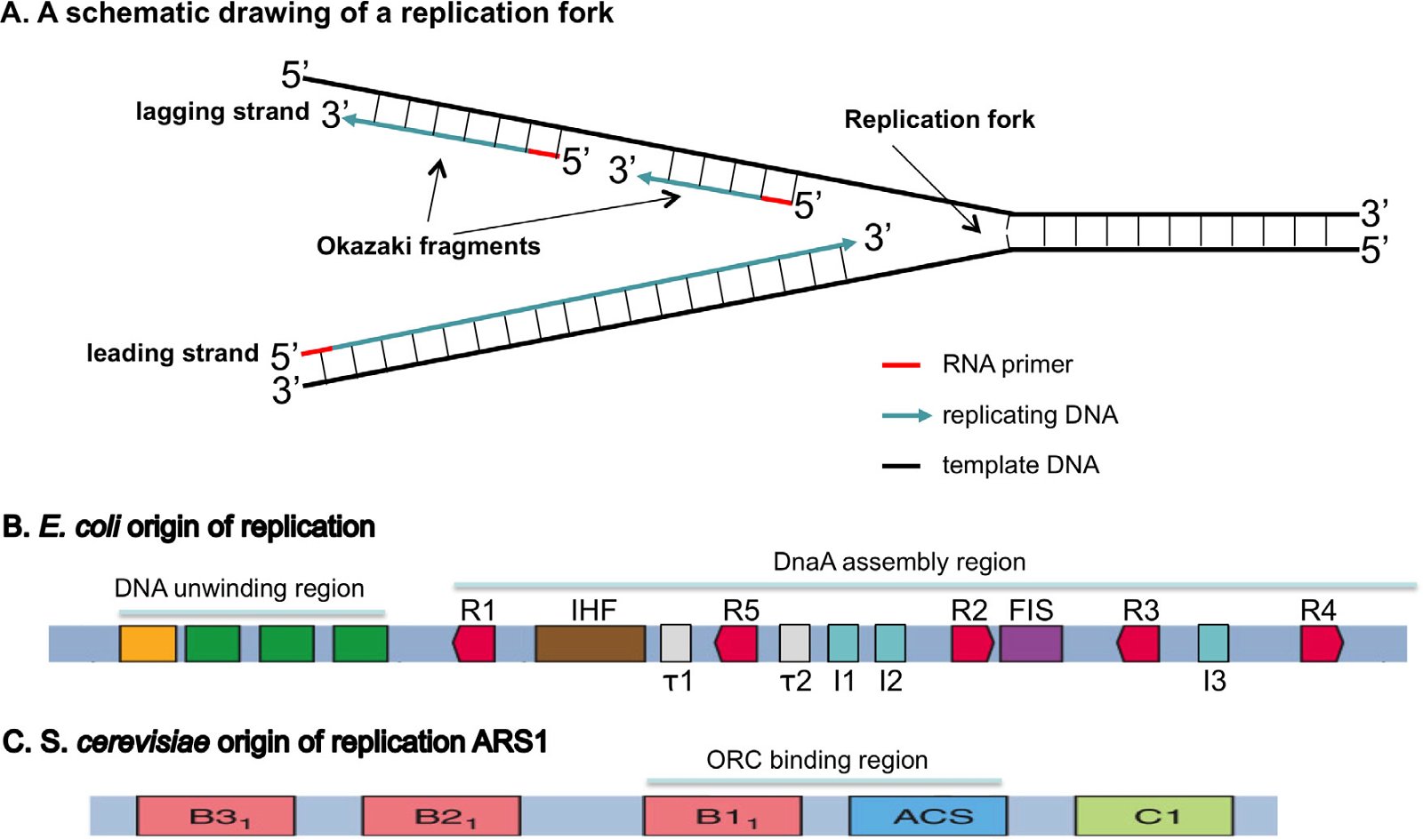
Fig. 3. Origins of replication. A: A schematic drawing of a replication fork. The double-stranded DNA will be unwound and each strand used as a template to build a complementary daughter strand during replication. All DNA replication starts with the synthesis of an RNA primer (red). The leading strand will be continuously synthesized, while the lagging strand is discontinuously synthesized as short Okazaki fragments. B: An E. coli origin of replication. The E. coli origin of replication contains a DNA unwinding region, which has three A/T rich 13 bp (13-mers, green) and an A/T rich cluster (orange), and a DnaA assembly region, which consists of ten 9-bp (9-mer) DnaA binding sites, including five DnaA boxes (R1-5, red), three I sites (I1–I3, blue), two τ sites (τ1 and τ2, white), a binding site for IHF (integration host factor, brown), and a binding site for FIS (factor for inversion stimulation, purple). IHF binding stimulates initiation, while FIS binding inhibits initiation. C: An S. cerevisiae origin of replication, autonomously replicating sequence 1 (ARS1). Yeast contains many origins of replication. These origins share an 11-bp element known as the A-element or the ARS consensus sequence (ACS). A mutation in the A region results in a complete loss of function. The A-element, however, is not sufficient for ARS function; specific 5′ and 3′ neighboring sequences are needed for ARS function. ARS1 has three B elements (B1, B2, and B3) and a C1 element. Different yeast ARSs may have different B elements and/or C elements. The Origin Recognition Complex (ORC) binds ARS1 by interacting with ACS and B1. Fig. 1B is modified from (Stepankiw et al. 2009) and 3C from Fig. 10.12 of Tropp (2012).
The leading strand of DNA is synthesized continuously, while the antiparallel lagging strand is synthesized discontinuously, as Okazaki fragments, because nucleotides are always added in a 5′ to 3′ direction in the cell. The RNA primers will be removed and exchanged with DNA by gap filling and the Okazaki fragments will be joined by DNA ligase. During the termination stage, terminators (in bacteria), or chromosomal ends called telomeres (in eukarya), will be synthesized. As detailed below, bacteria and eukarya differ in their requirements for DNA replication initiation and termination in both their DNA sequences (cis-elements) and in their trans-elements, i.e. the proteins that execute and regulate DNA replication (Forterre 2013; Leipe, Aravind, and Koonin 1999; Makarova and Koonin 2013; Merhej and Raoult 2012; O’Donnell, Langston, Stillman 2013; Skarstad and Katayama 2013).
A.1 Differences in the initiation site of replication
The first step of DNA replication is the recognition of start sites at the origins of replication by origin-binding proteins. Strikingly, the origins of DNA replication are species specific. Duplicating DNA in E. coli requires an E. coli-specific origin of replication, while duplication in S. cerevisiae requires an S. cerevisiae-specific origin of replication. These origins of replication are not interchangeable. This fact is experimentally demonstrated on a daily basis in many laboratories throughout the world: to clone and replicate an S. cerevisiae gene in E. coli requires a vector with an E. coli origin of replication, and to clone and replicate a bacterial gene in S. cerevisiae requires a vector carrying an S. cerevisiae origin of replication (plus a yeast centromere).
Bacteria and eukarya also differ in the numbers of origins of replication. Bacteria typically have a single circular chromosome with a single origin of replication, while eukarya have hundreds or thousands of origins of replication spread across multiple linear chromosomes. So how could evolution accomplish the change from one circular DNA strand to multiple linear chromosomes (for example, 16 pairs = 32 in yeast)? The bacterium might begin by evolving an enzyme to chop up its DNA into 16 pieces. But since it would have only one origin of replication, only 1/16th of the DNA would be replicated and the cell would die. It would have to simultaneously generate new origins of replication for all 16 pieces.
Furthermore, bacteria and eukarya differ in the definition of origins of replication, including the DNA sequence and the structure of the origin of replication. Bacterial origins of replication have well-defined sequences (Fig. 3B). In contrast, eukaryotic origins of replication in higher animals are typically not as clearly defined by specific DNA origin start site sequences, but by the organization of the chromatin—the DNA-protein-RNA compositional state of the chromosome (O’Donnell, Langston, and Stillman 2013). S. cerevisiae is the only known eukaryote that has specific sequence-defined origins of replication, though the sequence of S. cerevisiae origins of replication is completely different to that of bacteria (Fig. 3C). Logically one would expect that the difference of the origins of replication in sequence and/or structure in bacteria and eukarya requires different proteins to recognize them, which is indeed the case as will be discussed below.
A.2 Differences in core replication machineries
The key enzymes involved in bacterial and eukaryotic DNA replication differ dramatically in several major points [(Forterre 2013; Leipe, Aravind, and Koonin 1999), see Fig. 4 (E. coli on the left, and S. cerevisiae on the right)]:
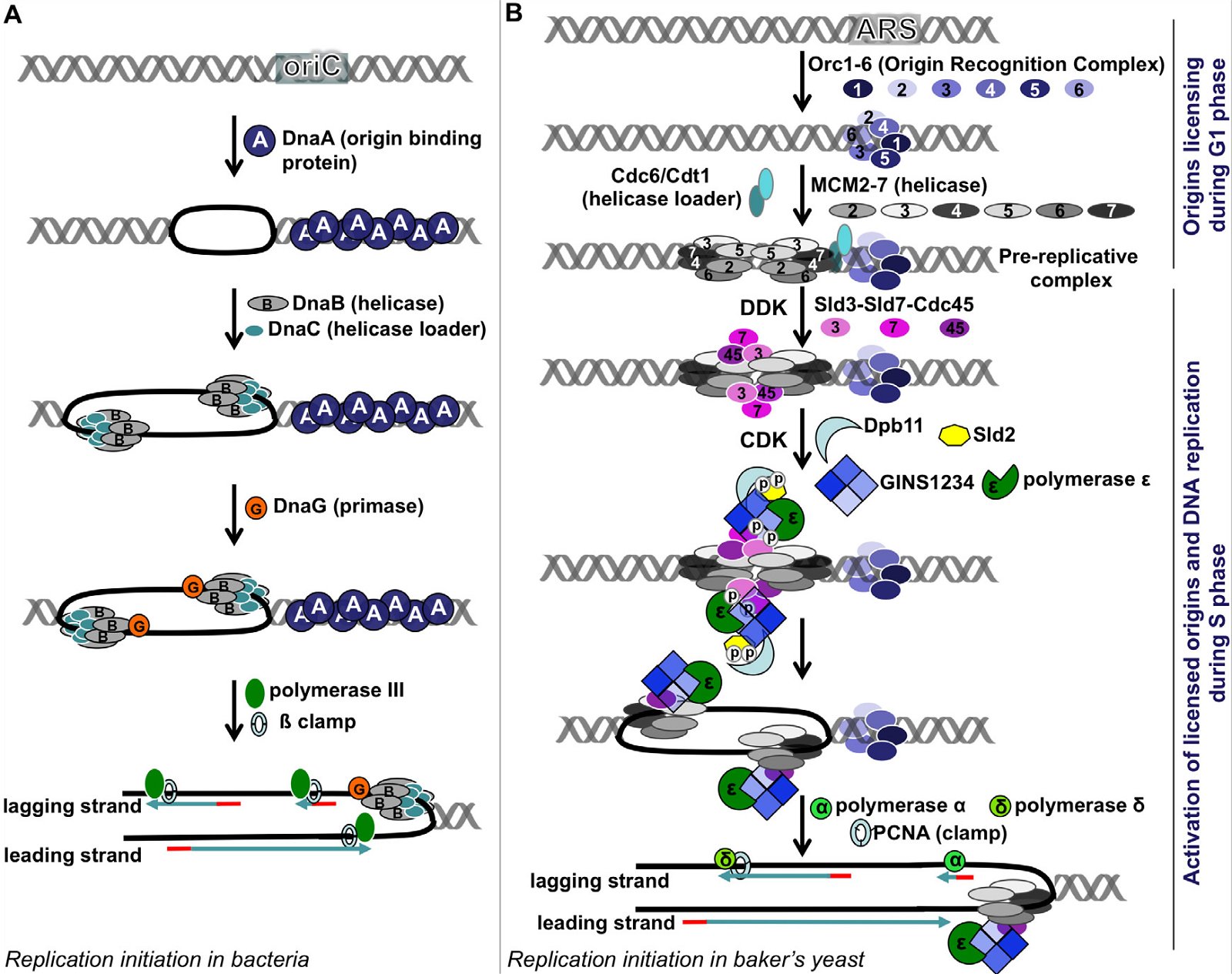
Fig. 4. A comparison of DNA replication initiation in bacteria E. coli and baker’s yeast S. cerevisiae. A: Initiation in E. coli. Multiple copies of DnaA bind the DnaA assembly region of oriC, resulting in melting of the DNA unwinding region, forming two replication forks. After that, a helicase loader DnaC loads helicase DnaB on the lagging strand, extending the single-stranded region. Recruitment of the primase followed by Polymerase III and ß clamp leads to synthesis of RNA primers and DNA. B: Initiation in yeast. The origins of replication are licensed during the G1 phase, starting with the binding of the ORC1-6 heterohexamer at the ARS. Subsequently, two copies of MCM 2-7 heterohexamers are loaded via Cdc6 and Cdt1 head to head onto double-stranded DNA, forming the pre-replication complex (pre-RC). When cells enter S phase, two cyclin-dependent kinases DDK and CDK phosphorylate Sld2, Sld3, and Sld7, enabling Sld3-Sld7-Cdc45 to bind the licensed origins by interacting with MCM2-7. Phosphorylation of Sld2 leads to the formation of a complex made of Dpb11, Sld2, heterotetrameric GINS protein, and Polymerase ε. This complex then joins the pre-replication complex, leading to the unwinding of the ARS and the formation of two replication forks. Each replication fork carries a CMG (Cdc45-MCM2-7-GINS) complex, which is the active helicase that unwinds DNA. The CMG is loaded on the leading strand, through a presently unknown mechanism. Subsequently, Polymerases α and δ and the DNA clamp PCNA are recruited and DNA replication begins. Yeast Pol α is composed of four subunits; its Pri1 subunit synthesizes RNA primers (8–12 nucleotides), its Pol1 subunit adds 15 to 25 deoxy-nucleotides to the primer, while the two other Pol α subunits regulate Pol1 and Pri1. After Pol α finishes making the initiating DNA, Pol δ and Pol ε come on board to synthesize the lagging strand and the leading strand, respectively. Fig. 2 is modified from Fig. 1 of (Li and Araki 2013). The order of arrangement of ORC1, ORC2, ORC3, ORC4, ORC5, and ORC6 in the ORC1-6 hexamer is according to (Tiengwe et al. 2012). Abbreviations: cdc: cell division cycle, CDK: cyclin-dependent kinase, Cdt1: chromatin licensing and DNA replication factor 1, DDK: Dbf4-dependent kinase, or Cdc7-Dbf4 kinase, Dpb11: Dpb = DNA Polymerase B, Dpb11 loads DNA Pol ε onto pre-replication complexes at origins of replication, GINS: Go-Ichi-Ni-San complex, MCM: mini-chromosome maintenance, ORC: Origin Recognition Complex, ori: Origin of replication, PCNA: Proliferating Cell Nuclear Antigen, Sld: synthetic lethality with dpb11.
First, in E. coli, a few copies of a single protein DnaA bind the DnaA assembly region of its origin of replication oriC (Figs. 3B and 4A). This causes melting of the A/T region of oriC. In contrast, the binding of the eukaryotic initiator, Origin Recognition Complex (ORC), is not able to unwind the DNA double helix, even though ORC is more complex and is made of six different proteins. How could an alleged evolving bacterium invent a complex eukaryotic ORC complex (assuming it already had generated origin of replication for all 16 pieces of the original circular DNA), if it could not unwind the DNA by itself, it would have certainly perished? And to complicate the issue further, the eukaryotic ORC only marks (licenses) the origin of replication. This licensing occurs during the G1 phase of the cell cycle, while the next step, activation of the origin of replication, occurs later, during the S phase, with the help of two S phase kinases DDK (Cdc7-Dbf4 kinase) and CDK (cyclin-dependent kinase). Whether or not a licensed origin will be used depends on the cell type and developmental or health status of a cell. More than a dozen proteins are required to activate a licensed origin. Consistent with the unique origins of replication in E. coli and S. cerevisiae, DnaA is non-existent in yeast or multicellular plants and animals, and the ORC and the many additional proteins required for the activation of eukaryotic origins of replication are not present in bacteria.
Second, no sequence similarity has been detected between bacterial and eukaryotic primases, their principle DNA polymerases, or their gap-filling polymerases (Aves, Liu, and Richards 2012; Costa, Hood and Berger 2013; Raymann et al. 2014).
Third, the DNA in eukaryotic chromosomes is wound up around a set of eight histone proteins that do not exist in bacteria—along with a complexly coded system of histone tail modifications that epigenetically control genes and genome function. Therefore, the eukaryotic DNA must be rearranged and precisely moved about for replication to begin and proceed.
Fourth, the DNA helicases that unwind the DNA for replication in bacteria and eukarya are loaded unto the opposite strands of DNA and moves in the opposite directions. The bacterial helicase, a homohexamer (contains six identical proteins), is loaded on the lagging strand and moves in the 5′ to 3′ direction of its bound DNA (Tuteja and Tuteja 2004). In contrast, the eukaryotic helicase, heterohexamer of six different proteins, is loaded on the leading strand and moves in the 3′ to 5′ direction of its bound DNA.
Finally, in bacteria, both the leading and lagging strands are synthesized by the same DNA polymerase. In eukarya, the two strands are synthesized by different polymerases, Pol ε for the leading strand and Pol δ for the lagging strand. Interestingly, neither Pol ε nor Pol δ can perform their function until another eukaryote-specific DNA polymerase Pol α, has added a certain number of deoxyribonucleotides to the RNA primers made by the primase required to initiate strand replication.
Not surprisingly, with the importance of DNA replication to the life of an organism, the vast majority of genes responsible for DNA replication are essential genes—their lack would render the organism inviable and unable to propagate its genome (see Fig. 2 of Tan and Tomkins [2015]).
Taken together, bacterial DNA replication machinery is not able even to initiate eukaryotic DNA replication. Thus, it is impossible for any bacteria to evolve into a eukaryote.
A.3 Differences in replication termination
Because of differences in their chromosomal structures, bacteria and eukarya face different issues at the completion of their chromosomal replication. For E. coli, with a circular genome, its DNA replication ceases at the termination sites, which contain special DNA sequences that are recognized by specific proteins named the terminus utilization substance. Things are very different for eukarya such as S. cerevisiae. Completely copying their linear chromosomes requires a complex ribonucleoprotein enzyme (contains multiple proteins plus RNA) that does not exist in bacteria, called telomerase (Tomkins and Bergman 2011). Without telomerase, the chromosomes of S. cerevisiae would be shortened during each round of cell division, and before long, chromosome ends would erode to the point of structural and functional failure. In other words, without first having the S. cerevisiae telomerase, which uses a yeast-specific chromosome end-cap RNA template (called a telomere repeat) to add telomeres to the chromosome ends, S. cerevisiae could not exist. Variants of this telomere-telomerase system are found in all eukarya. However, bacteria do not need this system so that they do not have it. A comparison of bacterial and eukaryotic replication can be found in Appendix A.
Therefore, not only is bacterial DNA replication machinery unable to initiate eukaryotic DNA replication, but it would also fail to complete the job even if it somehow magically succeeded in initiation. If a bacterium evolved into a yeast-like cell, and it somehow succeeded in passing new origins of replication onto all 16 chromosomes at the same time, they would need to create totally different origins of replication and all the proteins that can recognize the new origins of replication at about the same time. And of course all of them would have to be replicated into identical pairs to initiate the process of reproduction. At the same time, they would need to generate a centromere and all the other machinery needed to separate the pairs during cell division. This would require remarkable insight of evolution into the future, in producing protein parts for machines that do not yet exist and in creating machinery to assemble these machines that evolution realizes will be useful in the future.
How could a bacterium evolve into a eukaryote, which cannot use the bacterial machinery to copy its genome? The change from bacterium to eukaryote would inevitably entail the death of the organism since many essential genes would be lacking on either side of the gap. Likewise, the process of “devolving” a eukaryote into a bacterium would be just as lethal since the eukaryotic DNA replication machinery cannot be used to copy bacterial genomes. Similar analyses can be done with archaea (as we showed previously), another kind of prokaryote, with the same result, even though archaea replication machinery is more similar to that of eukarya than that of bacteria (Tan and Tomkins 2015). Therefore, there seems to be an evolutionarily impassable gap between prokaryotes and eukarya. The missing links between them will be forever missing because cell death is a barrier that the alleged mythical power of evolution cannot cross.
B. Transcription machines used by cells to transcribe their genomic genes
When pondering the origin of cellular machinery, it is not difficult for one to realize that despite its incredible complexity, DNA replication is the simplest of all the processes required for the engineering of the three main biopolymers, DNAs, RNAs, and proteins. As we shall see, more challenges to evolutionary theory are waiting when we consider transcription, and even more in translation. As was the case in DNA replication, during transcription and translation, prokaryotes differ dramatically from eukarya both in the initiation and termination stages. In addition, they also differ during the elongation steps of these two complex processes, but not as dramatically as during initiation and termination. Here we will just compare and contrast the bacterial and eukaryotic RNA polymerases and the processing of RNAs.
B.1 Differences in RNA polymerases
Eukarya have more RNA polymerases than bacteria and each eukaryotic RNA polymerase is made of more protein components than its bacterial counterpart. E. coli, like all other known prokaryotes, has a single RNA polymerase to synthesize all of its RNAs, including messenger RNA (mRNA), ribosomal RNA (rRNA), and transfer RNA (tRNA). The mRNAs are those that will be translated into proteins if they code for proteins. Many other mRNAs code for short and long noncoding RNAs that regulate gene and genome function. Ribosomal RNAs and tRNAs, with the help of many proteins, are responsible for the process of translating mRNAs into proteins. Protein-coding and noncoding RNA genes are very diverse, fall into many different categories, and generally are found in single, or low copy numbers, in the genome. In contrast, both rRNA genes and tRNA genes are high-copy number genes. For example, some plants contain thousands of copies of rRNA genes (Long and Dawid 1980).
S. cerevisiae, like all other eukarya, has three specialized RNA polymerases: Pol I for making the large ribosomal RNAs (rRNA), Pol II for making heteronuclear RNA (hnRNA), which are precursors of mRNA (pre-mRNA) and noncoding RNAs, and Pol III for producing small ribosomal RNA (5S rRNA), tRNAs, and other small RNAs. E. coli RNA polymerase is composed of five different protein subunits, while S. cerevisiae Pol I consists of 14 different proteins, Pol II has 12, and Pol III a total of 17. Most of the components of Pol I, II, and III have no bacterial counterparts (O’Donnell, Langston, and Stillman 2013).
Though being composed of more proteins, eukaryotic RNA polymerases are much less functionally independent than prokaryote RNA polymerases in the sense that the prokaryote RNA polymerases can transcribe their target genes by themselves or with very limited help from other proteins, while the eukaryotic RNA polymerases require the help of many different proteins and are unable to synthesize RNAs on their own. To transcribe a eukaryotic gene at the right level, at the right time, and at the right place, requires the involvement of many additional eukaryote-specific proteins, including transcription activators, mediators, and chromatin modifying and remodeling complexes. A comparison of bacterial and eukaryotic transcription can be found in Appendix B.
Part of the reason that eukarya require RNA polymerases different from prokaryote RNA polymerases is because eukaryotic genes and prokaryotic genes have different basic structures (Osbourn and Field 2009). Genes contain signatures unique to their own domains, especially in the promoter and terminator regions. A promoter is a special DNA sequence that, when recognized by transcription factors, dictates where and how much a gene will be transcribed, often with the help of other regulatory DNA sequences and their cognate protein binding partners. E. coli promoters and yeast promoters are distinct and cannot be cross recognized by their organism-specific transcription machinery. Even the single-subunit viral RNA polymerases are very specific (or particular) about what genes they will transcribe. For example, the single-subunit viral RNA polymerases Sp6 and T7, two polymerases commonly used in molecular biology laboratories for in vitro transcription, require a polymerase-specific promoter; the Sp6 RNA polymerase can only transcribe genes containing an Sp6 promoter, while the T7 RNA polymerase can only transcribe genes that have a T7 promoter. Therefore, one may not use the RNA polymerases from one domain, prokaryote or eukaryote, to transcribe genes from another domain because the RNA polymerases will only able to recognize the genes of its own domain of life, bacterial RNA polymerases mostly by themselves or with minimal help from other proteins, while eukaryotic RNA polymerases utilize the help of a sizeable set of eukaryotic-specific factors. Consequently, if one wishes to express a eukaryotic gene in bacteria, he/she must clone the gene of interest under a bacterial promoter in a bacterial vector that has a bacterial origin of replication. To express a bacterial gene in eukarya, you must clone the gene of interest under a eukaryotic promoter in a eukaryotic vector that has a eukaryotic origin of replication.
How could a prokaryote evolve into a eukaryote that is unable to use the prokaryotic RNA polymerase to transcribe the eukaryotic genome? And why would the eukaryotic polymerase arise first if it were useless in transcribing the prokaryotic genome?
B.2 Differences in RNA processing
Unlike mRNAs made in E. coli, which can be used directly to synthesize proteins, protein-coding genes in eukarya are transcribed in the nucleus as hetero-nuclear RNAs (hnRNAs or pre-mRNAs) that must be processed and exported from the nucleus to the cytoplasm for translation. In fact, in E. coli, a gene can be translated while it is still being transcribed. However, in eukarya, the products of the transcription machinery are only precursors of mRNAs that must be modified before they can be translated into proteins or used as noncoding RNAs for a variety of different cellular functions.
First, all eukaryote protein-coding and noncoding RNA gene transcripts, which are generated by Pol II, are capped with a 7-methylguanosine-modified nucleotide at their 5′ ends and most are also polyadenylated (poly A tail addition) at their 3′ ends (Hocine, Singer, and Grünwald 2010). Both modifications are necessary for the stability, subcellular localization/transport, and translation of the mRNAs that are protein coding. Interestingly, the 5′ caps in yeast are not the same as those found in multicellular organisms. The most complicated caps are found in vertebrates. While most mRNA in eukaryotes contains a poly-A tail, one unexpected exception for polyadenylation is that of histone pre-mRNAs and some types of long noncoding RNAs (Yi et al. 2013). Although histones do not exist in bacteria, they package and condense DNA into nucleosomes (like beads on a string) in eukarya and help establish the higher order structure of the chromatin. The histones of the nucleosomes also provide sites for chromatin modification to regulate gene and genome function. Histones are the most abundant housekeeping genes in eukarya, and most higher animal histone mRNAs do not have polyA tails. Bacterial RNAs are not capped at the 5′ end. Occasionally, bacteria add poly-A tails to their RNAs, but only to tag them for degradation, which is functionally opposite of the role played by eukaryote polyadenylation (Parton et al. 2014; Shandilya and Roberts 2012; Tropp 2012).
Second, all known eukarya contain genes with introns, although some eukarya have more intron-containing genes than others (Koonin, Csuros, and Rogozin 2013). It came as a surprise that the DNA of protein coding regions, named exons, of eukaryotic genes is often interrupted with non-protein coding regions, spliceosomal introns (referred to as introns hereafter), which are often much longer than exons. Intronic DNA is typically spliced out before the mRNA can be used to make proteins or long noncoding RNAs. Introns themselves often contain other genes for both short and long noncoding RNAs, in addition to many other chromosomal signals required for gene and genome function (discussed more below).
Intron splicing is a very demanding process. Specific DNA sequences are required at the 5′ end, 3′ end, and inside of each intron, known as the 5′ splicing site or the splicing donor, the 3′ splicing site or the splicing acceptor, and the branching point, respectively. Occasionally, splicing enhancers or silencers inside exons or introns are also necessary to ensure correct intron-exon recognition and splicing. Furthermore, many specialized small RNAs and proteins are required for intron splicing. The human spliceosome is composed of at least 170 proteins and five small nuclear RNAs (Behzadnia et al., 2007). Thus, intronic sequences are not random junk DNA as some have thought. On the contrary, introns are essential for the normal operation and survival of eukarya. Animals use highly regulated splicing to deal with stress and even to determine sex identity (Biamonti and Caceres 2009; David and Manley 2008; Matlin, Clark, and Smith 2005; Sosnowski, Belote, and McKeown 1989). Misregulation of splicing is the underlying cause of many human genetic disorders (Matlin, Clark, and Smith 2005; Tang et al. 2013). Deletion of intron sequence may even cause lethal diseases in humans (Szafranski et al. 2013)
Thus, all eukaryotes have RNAs need to be capped, polyadenylated, or spliced—three major features that do not exist in bacteria. Successful capping, poly-A tail addition, and intron-splicing require many eukaryotic-specific genes. Without the function of these essential systems, no eukaryote can exist. The dilemma is that the many genes encoding the corresponding cellular machinery are not found in bacteria because bacteria do not need them.
C. Translation machines used by cells to translate their protein-coding genes
Just as we have seen with respect to the machinery for DNA replication and transcription, the molecular complexes used for translation in bacteria and yeast share a few basic designs, but they are not interchangeable and the differences are too vast to be explained by evolution.
C.1 Similarity in basic design
The machines that translate mRNA into proteins are called ribosomes. All ribosomes are made of two subunits (Tropp 2012). The large ribosome subunit houses the peptide bond-forming center while the small subunit decodes the message carried by mRNA. The go-between translators are transfer RNA (tRNA). One end of a tRNA interacts with the large ribosomal subunit in the language of proteins while the other end interacts with mRNA in the language of nucleic acids. Both prokaryotic ribosomes and eukaryotic ribosomes are composed of ribosomal RNA (rRNA) and ribosomal proteins. However, as discussed below, prokaryotic ribosomes can only translate prokaryotic transcripts into proteins, and eukaryotic ribosomes can only translate eukaryotic transcripts. Thus, to express a eukaryotic protein in a bacteria artificially in the laboratory, the eukaryotic protein-coding gene needs to be cloned into a bacterial vector, so that the eukaryotic gene can be transcribed and translated by the bacterial machinery. In addition, if the eukaryotic gene contains introns, the process will not work. You must use a DNA copy of a processed mRNA that has all of its introns removed.
C.2 The critical differences
We will describe two categories of differences between bacteria and eukaryotes, one is compositional and the other functional below:
C.2.1 The non-interchangeable components
1. Differences in Mature Ribosomes
E. coli ribosomes are made of three types of rRNAs: 5S rRNA, 23S rRNA, and 16S rRNA, along with 55 ribosomal proteins (Korobeinikova, Garber, and Gongadze 2012). The 5S rRNA, 23S rRNA, and 33 proteins together form the large subunit, while 16S rRNA and 22 other proteins form the small subunit. Of the 55 E. coli ribosomal proteins, 45 have experimentally been shown to be essential for the viability of E. coli (Baba et al. 2006; Gerdes et al. 2003; Lecompte et al. 2002; Zhang and Lin 2009). A total of 57 ribosomal proteins have been found in bacteria, 23 of which are unique to bacteria (22 in E. coli); they are not found in eukarya (Korobeinikova, Garber, and Gongadze 2012; Lecompte et al. 2002).
Eukaryotic ribosomes are much more complicated than bacterial ribosomes, with four (instead of the three in bacteria) ribosomal RNAs and ~78 (instead of ~57 as in bacteria) ribosomal proteins (Tropp 2012). The large ribosomal subunit of S. cerevisiae contains three types of rRNA: 5S, 5.8S, and 28S rRNAs, as well as 46 proteins. The small ribosomal subunit contains one rRNA (18S rRNA) and 32 proteins. Forty-three of the 78 S. cerevisiae ribosomal proteins have no bacterial homologs (Lecompte et al. 2002). Nineteen S. cerevisiae ribosomal genes have been experimentally shown to be essential for the viability of S. cerevisiae, even though five of the 19 essential ribosomal genes have sister genes, known as paralogs, with almost the same protein-coding regions (Giaever et al. 2002; Lecompte et al. 2002; Zhang and Lin 2009).
2. Differences in Ribosome Assembly Factors
Hundreds of “behind-the-scenes” heroes are involved in facilitating the protein synthesis that goes on in the mature ribosomes. For example, more than 150 non-ribosomal proteins and more than 70 non-protein coding RNAs function in a hierarchical manner to assemble the yeast ribosome (Henras et al. 2008; Kressler, Hurt, and Bassler 2010). Several hundred proteins have been implicated in human ribosomal biogenesis (Andersen et al. 2002; Andersen et al. 2005; Couté et al. 2006; Moss et al. 2007; Scherl et al. 2002). Although things are much simpler in E. coli, more than 20 assembly factors, including helicases, GTPases, and chaperones, are necessary for assembling E. coli ribosomes (Chen and Williamson 2013; Shajani, Sykes, and Williamson 2011).
3. Differences in Ribosomal RNA Genes (rDNAs)
Perhaps surprisingly, the ribosomal RNA genes (rDNAs) differ significantly among different organisms, even though their coding products (rRNAs) perform the same essential role of synthesizing proteins (with the help of their binding protein partners) (Fig. 5A). The rDNAs cannot be interchanged even between alleged closely related animal species (Pikaard 2002). For example, mouse rDNAs cannot be transcribed with the human transcription machinery, and vice versa. A close analysis of rDNAs shows that the coding regions of rDNAs, the regions that will be transcribed into rRNA, have similar sequences even across very diverse organisms, although there are species-specific extensions (Fig. 5B). However, the non-coding regions determine whether and how much rDNA will be expressed, and these vary greatly in different species, both in length and in DNA sequence (Fig. 5A and [Pikaard 2002]). Consequently, rDNA transcription is often species-specific and requires species-specific transcription factors (Pikaard 2002).
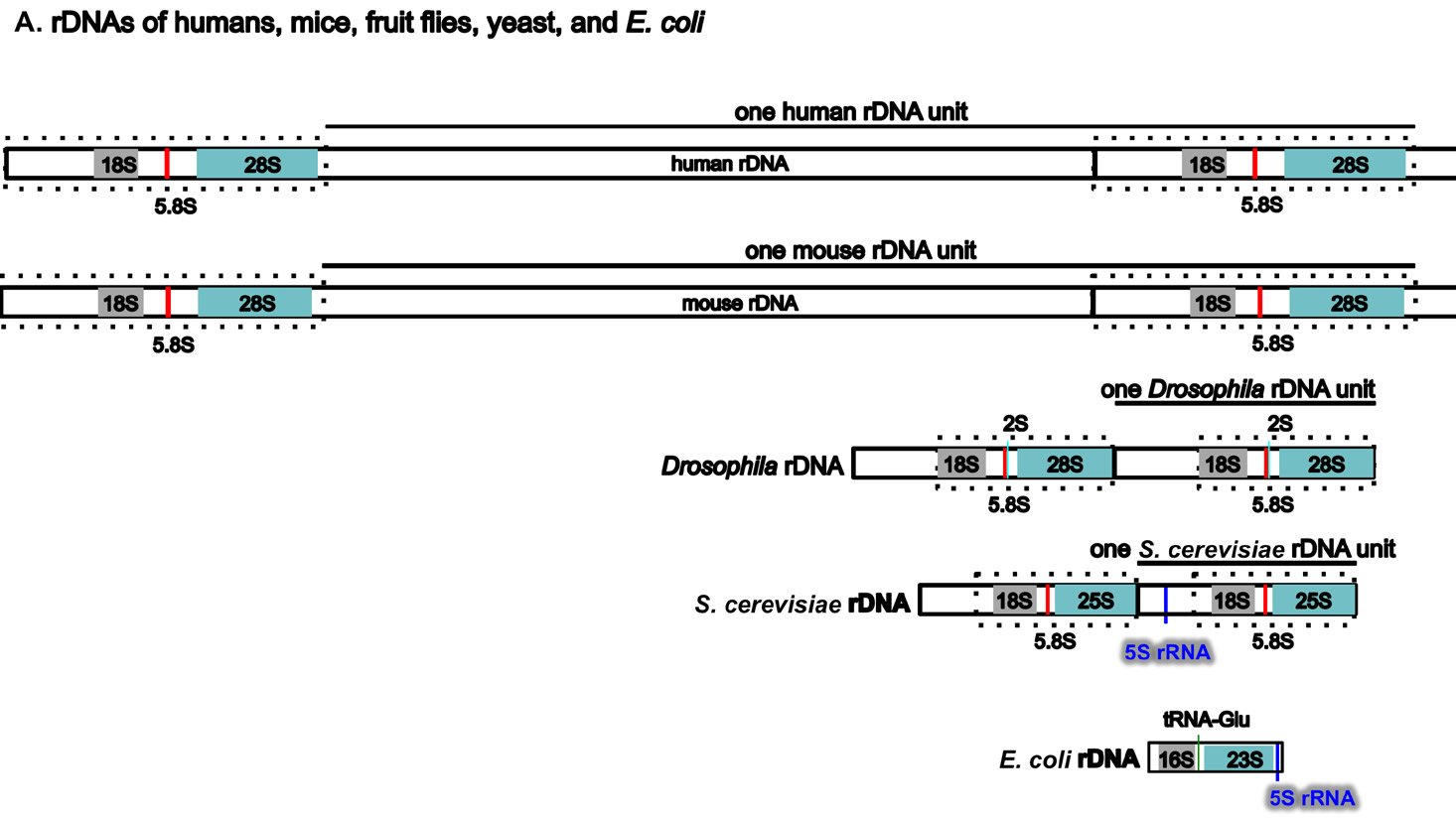
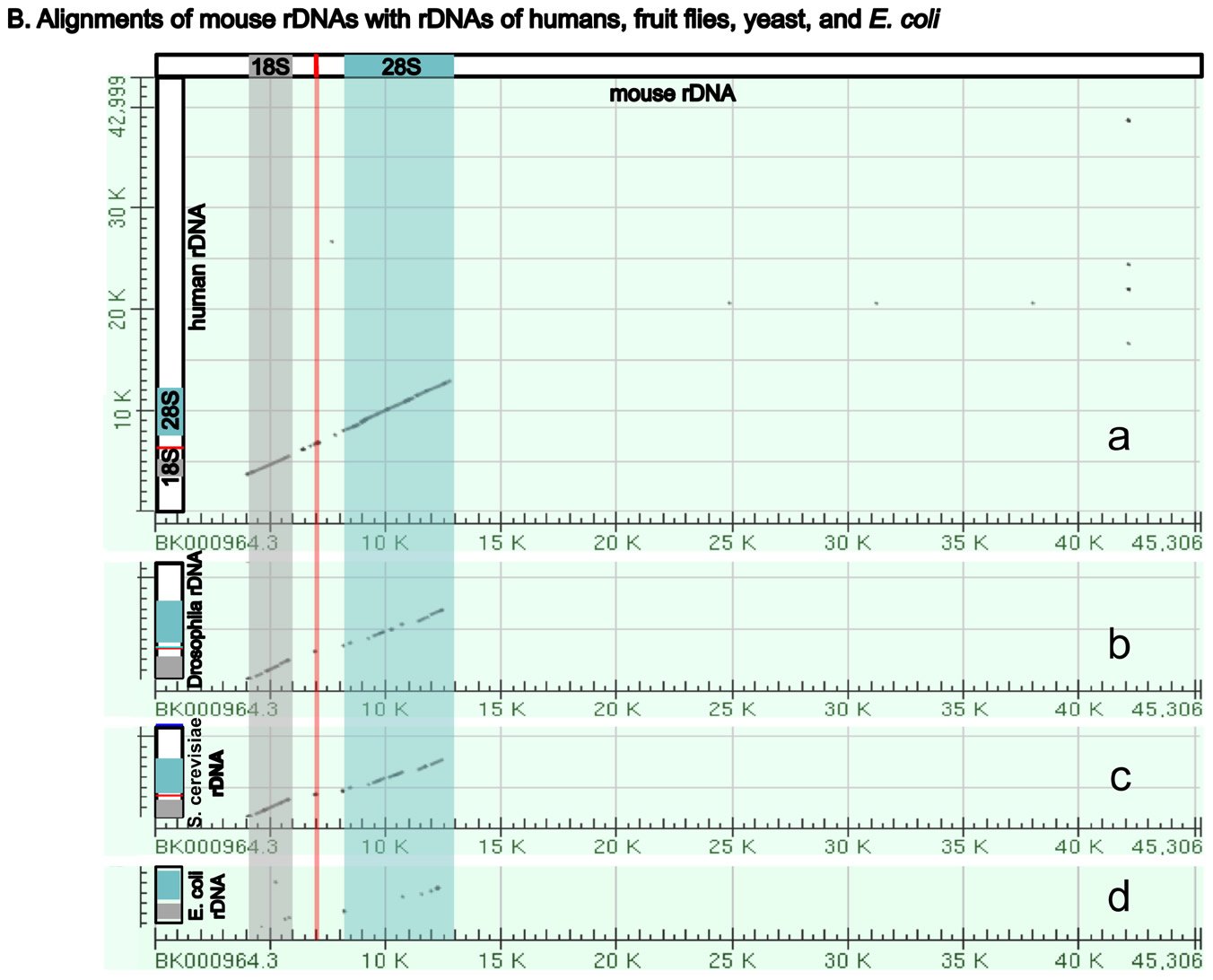
Fig. 5. A comparison of the rDNAs of humans, mice, Drosophila, yeast and E. coli. A. A schematic drawing of human, mouse, Drosophila, yeast and E. coli rDNA. Eukaryotic rDNAs are high copy number genes. Each copy contains a transcribed region (boxed with dashed lines), which is transcribed into pre-rRNA, and a regulatory non-transcribed region, which contains the information (promoters, enhancers, etc.) to control the transcription of the pre-rRNA. The pre-rRNAs are subsequently processed by endo- and exo-nucleases into mature rRNA (mature 18S rRNAs in light gray, 28S rRNAs in blue, and 5.8S rRNAs in red). Yeast rDNA differs from the rDNAs of other eukaryotes in that a 5S rRNA gene is sandwiched in between two rDNA transcribed units. Interestingly, the 5S rRNA gene is transcribed from the opposite direction than the pre-rRNA. Drosophila rDNA contains an insect-specific 2S rRNA (green, next to the 5.8S rRNA). The E. coli counterparts of eukaryotic18S and 28S rRNA are 16S and 23S rRNA, respectively. Bacteria do not have any 5.8S counterpart. E. coli rDNA is organized into operons, each of which codes not only 16S rRNA and 23S rRNA, but also 5S rRNA and tRNA. All genes in an operon will be transcribed as a single unit. Consequently, E. coli 16S rRNA, 23S rRNA, 5S rRNA, and tRNA in the same operon are transcribed as one single unit and are later processed by nucleases into mature rRNAs and tRNAs. Different operons contain different numbers and/or kinds of tRNA. All rDNAs are drawn to the same scale. B. Alignments of mouse rDNA (X-axis) with the rDNAs of (a) humans, (b) Drosophila, (c) yeast, and (d) E. coli (Y-axes). All Y-axes have the same scale and only that for the human is labeled. Note that the regions corresponding to mature rRNA are highly conserved from yeast to humans (a-c). Similarity can be readily detected even between mouse 18S rRNA and E. coli 16S rRNA, and between mouse 28S rRNA and E. coli 23S rRNA (d). In contrast, the control regions are very diverse, even between the rDNA of humans and mice. Sequences of rDNA are from NCBI (The GenBank IDs are: mouse Mus musculus-BK000964.3, human Homo sapiens-U13369.1, D. melanogaster- M21017.1, yeast S. cerevisiae-U53879.1, E. coli-AB035921.1). Pair-wise sequence comparisons were performed using Gapped BLAST and PSI-BLAST (Altschul et al. 1997), using the parameter “somewhat similar sequences” (blastn) on the NCBI BLAST website http://blast.ncbi.nlm.nih.gov.
Three features of rDNAs are puzzling to the evolutionist, especially when considered together. First, they are depicted by high copy-number genes, constituting hundreds to thousands of copies (Long and Dawid 1980). Second, all the copies within an organism are almost identical. Third, ribosomal genes between different organisms are highly species-specific in that rDNAs from one species cannot be transcribed by its close relatives. Some suggest that a mythical process called concerted evolution accounts for the high homogeneity within a species and the dramatic differences between species (Eickbush and Eickbush 2007; Naidoo et al. 2013; Nei and Rooney 2005). According to this view, all the rDNAs in an organism magically change together in unison, then suddenly, a new species is born. Proponents of this idea seem to forget about the many proteins that are required to transcribe the rDNAs, those which cannot function at all except in transcribing their own species-specific rDNAs. In addition, they tend to forget that genes are all linked in one or a few chromosomes. Genes are not like independent units that can freely move within the chromosome or be freely and independently changed around the genome. Differences in ribosomal RNAs are also accompanied by differences in ribosomal binding proteins (Roberts et al. 2008). In other words, not only rDNAs, but also the rRNA-associated ribosomal proteins, species-specific rDNA transcription factors, the rRNA modifying factors, and ribosomal assembling factors in an organism must change in a miraculous coordinated fashion for evolution to be feasible. All of these have to occur at once; small independent changes would be lethal. But how would evolution make all these changes simultaneously? It would have to plan ahead very carefully or the death of the cells involved would hinder its progress significantly, but of course evolution is a random mindless naturalistic process incapable of strategizing or planning anything.
4. Differences in Ribosomal Protein Genes
As mentioned, many ribosomal proteins are unique to their domains in the schema of life. Twenty-three of the 57 bacterial ribosomal proteins are only found in bacteria and 43 of the 78 yeast ribosomal proteins do not have bacterial counterparts. In addition, eukaryotic ribosomal protein genes have introns, which are not found in bacteria. Indeed, introns seem to be enriched in ribosomal protein genes. For example, only 2.2 % of the non-ribosomal protein genes in yeast contain introns but about 74% of yeast ribosomal protein genes have introns (Ares, Grate, and Pauling 1999; Parenteau et al. 2011). As discussed above, intron splicing is a demanding task; to splice an intron out of its pre-mRNAs requires more than a hundred molecules, including proteins and RNAs. However, bacteria do not have genes required for intron splicing. Therefore, eukaryotic ribosomal protein genes cannot be processed in bacteria. Furthermore, 59 of the 78 S. cerevisiae ribosomal proteins are each encoded by two genes, so-called paralogs, with almost identical protein-coding regions, and thus S. cerevisiae has 137 ribosomal genes. Contrary to what might be expected for two highly similar genes, these paralogous ribosomal proteins are functionally distinct (Komili et al. 2007). Thus, to summarize, bacterial ribosomes can only be made in bacteria and eukaryotic ribosomes only in eukarya not only due to their unique ribosomal proteins but also because of the specialized complexity required in eukarya for intron splicing and ribosome assembly.
C.2.2 Ribosomal function is not exchangeable between bacteria and yeast
Due to differences in composition between bacterial and yeast ribosomes, bacterial ribosomes can only translate bacterial transcripts, and eukaryotic ribosomes can only translate eukaryotic transcripts (Connolly and Culver 2009; Korobeinikova, Garber, and Gongadze 2012; Panse and Johnson 2010; Shajani, Sykes, and Williamson 2011; Strunk and Karbstein 2009). Translation start sites in many E. coli genes are determined by the exact base-pairing of a special region, the Shine-Dalgarno sequence, and the complementary sequence in the 16S rRNA. Eukarya do not use the Shine-Dalgarno sequence to determine where to start translation. Instead, eukarya use the 5′ cap and the poly-A tail, via a so-called scanning model of initiation, to determine the direction of translation and often start its translation at the 5′-most start codon. Some eukaryotic genes contain a sequence called a Kozak sequence, including bases -3 to +1 where +1 is the first base in the start codon, to facilitate their correct translation. Kozak sequences are useless in facilitating bacteria translation. Therefore, to express a eukaryotic protein in bacteria, the eukaryotic protein-coding gene has to be cloned into a bacterial vector with a bacterial promoter, so that the eukaryotic gene can be transcribed artificially with the Shine-Dalgarno sequence that is necessary for it to be translated by the bacterial machinery. In contrast, to express a bacterial gene in a eukaryotic cell, the bacterial gene has to be cloned into a eukaryotic vector with a eukaryotic promoter, so that the bacterial gene can be transcribed with a 5′ cap and a poly-A tail and be recognized by the eukaryotic ribosomes. A comparison of bacterial and eukaryotic translation can be found in Appendix C.
Conclusions
In summary, evolutionists are faced with a dilemma: on the one hand, all life forms share common basic building materials and many biological processes. On the other hand, there are many unbridgeable gaps between diverse organisms, as shown by the many indispensable and non-interchangeable molecular machines that replicate, transcribe, and translate the genetic code. Attempts to find one single gene tree to depict the relationships of different life forms have failed (reviewed by Tomkins and Bergman 2013). However, giving up on one tree does not mean we must accept the mess of a tangled web as some have suggested (Bapteste et al. 2013; Suárez-Diaz and Anaya-Muñoz 2008). A forest of trees is more likely to represent the real history of life, as indicated by several studies by Koonin and colleagues (Koonin 2007, 2009; Koonin, Wolf, and Puigbo 2009; Koonin, Puigo, and Wolf 2011; Puigbo, Wolf, and Koonin 2009, 2012, 2013). This observation fits well with the creationist prediction of an orchard of life, not a tree of life (Tomkins and Bergman, 2013). Therefore, the current knowledge of the molecular details of life is consistent with the creation record of the Bible in that all animals, including humans, were each made “according to its kind.”
Acknowledgments
We are grateful to Casey Luskin (Discovery Institute, Seattle, WA) and Richard Swindell (Columbia, Missouri) for their critical reading of the manuscript and constructive feedback.
References
Alberts, B. 1998. The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 92, no. 3:291–294.
Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25, no. 17:3389–3402.
Andersen, J. S., Y. W. Lam, A. K. Leung, S. E. Ong, C. E. Lyon, A. I. Lamond, and M. Mann. 2005. Nucleolar proteome dynamics. Nature 433, no. 7021:77–83. doi: 10.1038/nature03207.
Andersen, J. S., C. E. Lyon, A. H. Fox, A. K. Leung, Y. W. Lam, H. Steen, M. Mann, and A. I. Lamond. 2002. Directed proteomic analysis of the human nucleolus. Current Biology 12, no. 1:1–11.
Ares, M. Jr., L. Grate, and M. H. Pauling. 1999. A handful of intron-containing genes produces the lion’s share of yeast mRNA. RNA 5, no. 9:1138–1139.
Aves, S. J., Y. Liu, and T. A. Richards. 2012. Evolutionary diversification of eukaryotic DNA replication machinery. Subcell Biochemistry 62:19–35. doi: 10.1007/978-94-007-4572-8_2.
Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology 2:2006 0008. doi: 10.1038/msb4100050.
Bapteste, E., L. van Iersel, A. Janke, S. Kelchner, S. Kelk, J. O. McInerney, D. A Morrison, et al. 2013. Networks: expanding evolutionary thinking. Trends in Genetics 29, no. 8:439–441. doi: 10.1016/j.tig.2013.05.007.
Behzadnia, N., M. M. Golas, K. Hartmuth, B. Sander, B. Kastner, J. Deckert, P. Dube, et al. 2007. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO Journal 26, no. 6:1737–1748.
Biamonti, G., and J. F. Caceres. 2009. Cellular stress and RNA splicing. Trends in Biochemical Sciences 34, no. 3:146–153. doi: 10.1016/j.tibs.2008.11.004.
Chen, S. S, and J. R Williamson. 2013. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. Journal of Molecular Biology 425, no. 4:767–779. doi: 10.1016/j.jmb.2012.11.040.
Chen, W. H., P. Minguez, M. J. Lercher, and P. Bork. 2012. OGEE: an online gene essentiality database. Nucleic Acids Research 40 (Database issue):D901-6. doi: 10.1093/nar/gkr986.
Connolly, K., and G. Culver. 2009. Deconstructing ribosome construction. Trends in Biochemical Sciences 34, no. 5:256– 263. doi: 10.1016/j.tibs.2009.01.011.
Costa, A., I. V. Hood, and J. M. Berger. 2013. Mechanisms for initiating cellular DNA replication. Annual Review of Biochemistry 82:25–54. doi: 10.1146/annurev-biochem-052610-094414.
Couté, Y., J. A. Burgess, J. J. Diaz, C. Chichester, F. Lisacek, A. Greco, and J. C. Sanchez. 2006. Deciphering the human nucleolar proteome. Mass Spectrometry Reviews 25, no. 2:215–234. doi: 10.1002/mas.20067.
Darwin, C. 1859. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. London, United Kingdom: John Murray.
David, C. J., and J. L. Manley. 2008. The search for alternative splicing regulators: new approaches offer a path to a splicing code. Genes & Development 22, no. 3:279–285. doi: 10.1101/gad.1643108.
Ebersberger, I., P. Galgoczy, S. Taudien, S. Taenzer, M. Platzer, and A. von Haeseler. 2007. Mapping human genetic ancestry. Molecular Biology and Evolution 24, no. 10:2266–2276. doi: 10.1093/molbev/msm156.
Eickbush, T. H., and D. G. Eickbush. 2007. Finely orchestrated movements: Evolution of the ribosomal RNA genes. Genetics 175, no. 2:477–485. doi: 10.1534/genetics.107.071399.
Forterre, P. 2013. Why are there so many diverse replication machineries? Journal of Molecular Biology 425, no. 23:4714– 4726. doi: 10.1016/j.jmb.2013.09.032.
Gerdes, S. Y., M. D. Scholle, J. W. Campbell, G. Balazsi, et al. 2003. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. Journal of Bacteriology 185, no. 19:5673–5684.
Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Véronneau, S. Dow. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, no. 6896:387–391. doi: 10.1038/nature00935.
Glass, J. I., N. Assad-Garcia, N. Alperovich, S. Yooseph, M. R. Lewis, M. Maruf, C. A. Hutchison III, H. O. Smith and J. C. Venter. 2006. Essential genes of a minimal bacterium. Proceedings of the National Academy of Sciences U.S.A 103, no. 2:425–430. doi: 10.1073/pnas.0510013103.
Henras, A. K., J. Soudet, M. Gérus, S. Lebaron, M. Caizerques-Ferrer, A. Mougin, and Y. Hentry. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cellular and Molecular Life Sciences 65, no. 15:2334–2359. doi: 10.1007/s00018-008-8027-0.
Hocine, S., R. H. Singer, and D. Grünwald. 2010. RNA processing and export. Cold Spring Harbor Perspectives in Biology 2, no. 12:a000752. doi: 10.1101/cshperspect.a000752.
Komili, S., N. G Farny, F. P Roth, and P. A Silver. 2007. Functional specificity among ribosomal proteins regulates gene expression. Cell 131, no. 3:557–571. doi: 10.1016/j. cell.2007.08.037.
Koonin, E. V. 2007. The Biological Big Bang model for the major transitions in evolution. Biology Direct 2:21. doi: 10.1186/1745-6150-2-21.
Koonin, E. V. 2009. Darwinian evolution in the light of genomics. Nucleic Acids Research 37, no. 4:1011–1034. doi: 10.1093/nar/gkp089.
Koonin, E. V., M. Csuros, and I. B. Rogozin. 2013. Whence genes in pieces: reconstruction of the exon-intron gene structures of the last eukaryotic common ancestor and other ancestral eukaryotes. Wiley Interdisciplinary Reviews RNA 4, no. 1:93–105. doi: 10.1002/wrna.1143.
Koonin, E. V., P. Puigbo, and Y. I. Wolf. 2011. Comparison of phylogenetic trees and search for a central trend in the “forest of life”. Journal of Computational Biology 18, no. 7:917–924. doi: 10.1089/cmb.2010.0185.
Koonin, E. V., Y. I. Wolf, and P. Puigbo. 2009. The phylogenetic forest and the quest for the elusive tree of life. Cold Spring Harbor Symposia on Quantitative Biology 74:205–213. doi: 10.1101/sqb.2009.74.006.
Korobeinikova, A. V., M. B. Garber, and G. M. Gongadze. 2012. Ribosomal proteins: structure, function, and evolution. Biochemistry (Moscow) 77, no. 6:562–574. doi: 10.1134/S0006297912060028.
Kressler, D., E. Hurt, and J. Bassler. 2010. Driving ribosome assembly. Biochimica et Biophysica Acta 1803, no. 6:673– 683. doi: 10.1016/j.bbamcr.2009.10.009.
Kumar, S., and S. B. Hedges. 2011. TimeTree2: species divergence times on the iPhone. Bioinformatics 27, no. 14:2023–2024. doi: 10.1093/bioinformatics/btr315.
Lecompte, O., R. Ripp, J. C. Thierry, D. Moras, and O. Poch. 2002. Comparative analysis of ribosomal proteins in complete genomes: an example of reductive evolution at the domain scale. Nucleic Acids Research 30, no. 24:5382–5390.
Leigh, J. W., F. J. Lapointe, P. Lopez, and E. Bapteste. 2011. Evaluating phylogenetic congruence in the post-genomic era. Genome Biology and Evolution 3:571–587. doi: 10.1093/gbe/evr050.
Leipe, D. D, L Aravind, and E. V Koonin. 1999. Did DNA replication evolve twice independently? Nucleic Acids Research 27, no. 17:3389–3401.
Li, Y., and H. Araki. 2013. Loading and activation of DNA replicative helicases: the key step of initiation of DNA replication. Genes to Cells 18, no. 4:266–277. doi: 10.1111/gtc.12040.
Liao, B-Y., and J. Zhang. 2008. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proceedings of the National Academy of Sciences U.S.A. 105, no. 19:6987–6992. doi: 10.1073/pnas.0800387105.
Liao, B-Y., and J. Zhang. 2007. Mouse duplicate genes are as essential as singletons. Trends in Genetics 23, no. 8:378– 381. doi: 10.1016/j.tig.2007.05.006.
Long, E. O., and I. B. Dawid. 1980. Repeated genes in eukaryotes. Annual Review of Biochemistry 49:727–764. doi: 10.1146/annurev.bi.49.070180.003455.
Makarova, K. S., and E. V Koonin. 2013. Archaeology of eukaryotic DNA replication. Cold Spring Harbor Perspectives in Biology 5, no. 11:a012963.
Matlin, A. J., F. Clark, and C. W. Smith. 2005. Understanding alternative splicing: towards a cellular code. Nature Reviews: Molecular Cell Biology 6, no. 5:386–398. doi: 10.1038/nrm1645.
Merhej, V., and D. Raoult. 2012. Rhizome of life, catastrophes, sequence exchanges, gene creations, and giant viruses: how microbial genomics challenges Darwin. Frontiers in Cellular and Infection Microbiology 2:113. doi: 10.3389/fcimb.2012.00113.
Moss, T., F. Langlois, T. Gagnon-Kugler, and V. Stefanovsky. 2007. A housekeeper with power of attorney: the rRNA genes in ribosome biogenesis. Cellular and Molecular Life Sciences 64, no. 1:29–49. doi: 10.1007/s00018-006-6278-1.
Naidoo, K., E. T. Steenkamp, M. P. A. Coetzee, M. J. Wingfield, and B. D. Wingfield. 2013. Concerted evolution in the ribosomal RNA cistron. PLoS One 8, no. 3:e59355. doi: 10.1371/journal.pone.0059355.
Nei, M., and A. P. Rooney. 2005. Concerted and birth-and-death evolution of multigene families. Annual Review of Genetics 39:121–152. doi: 10.1146/annurev.genet.39.073003.112240.
NSF. 2010. Assembling the Tree of Life (ATOL), NSF 10-513 NSF.
O’Donnell, M., L. Langston, and B. Stillman. 2013. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harbor Perspectives in Biology 5, no. 7. doi: 10.1101/cshperspect.a010108.
Osbourn, A. E., and B. Field. 2009. Operons. Cellular and Molecular Life Sciences 66, no. 23:3755–3775. doi: 10.1007/s00018-009-0114-3.
Page, R. D. 2012. Space, time, form: viewing the Tree of Life. Trends in Ecology and Evolution 27, no. 2:113–120. doi: 10.1016/j.tree.2011.12.002.
Panse, V. G., and A. W. Johnson. 2010. Maturation of eukaryotic ribosomes: acquisition of functionality. Trends in Biochemical Sciences 35, no. 5:260–266. doi: 10.1016/j. tibs.2010.01.001.
Parenteau, J., M. Durand, G. Morin, J. Gagnon, J-F. Lucier, R. J. Wellinger, B. Chabot, and S. A. Elela. 2011. Introns within ribosomal protein genes regulate the production and function of yeast ribosomes. Cell 147, no. 2:320–331. doi: 10.1016/j.cell.2011.08.044.
Parton, R. M, A. Davidson, I. Davis, and T. T Weil. 2014. Subcellular mRNA localisation at a glance. Journal of Cell Science 127, no. 10:2127–2133. doi: 10.1242/jcs.114272.
Penel, S., A-M. Arigon, J-F. Dufayard, A-S. Sertier, V. Daubin, L. Duret, M. Gouy, and G. Perrière, G. 2009. Databases of homologous gene families for comparative genomics. BMC Bioinformatics 10 Suppl 6, S3.
Pikaard, C. S. 2002. Transcription and tyranny in the nucleolus: the organization, activation, dominance and repression of ribosomal RNA genes. The Arabidopsis Book 1:e0083. doi: 10.1199/tab.0083.
Puigbo, P., Y. I. Wolf, and E. V. Koonin. 2009. Search for a ‘Tree of Life’ in the thicket of the phylogenetic forest. Journal of Biology 8, no. 6:59. doi: 10.1186/jbiol159.
Puigbo, P., Y. I. Wolf, and E. V. Koonin. 2012. Genome-wide comparative analysis of phylogenetic trees: the prokaryotic forest of life. Methods in Molecular Biology 856:53–79. doi: 10.1007/978-1-61779-585-5_3.
Puigbo, P., Y. I. Wolf, and E. V. Koonin. 2013. Seeing the Tree of Life behind the phylogenetic forest. BMC Biology 11:46. doi: 10.1186/1741-7007-11-46.
Raymann, K., P. Forterre, C. Brochier-Armanet, and S. Gribaldo. 2014. Global phylogenomic analysis disentangles the complex evolutionary history of DNA replication in Archaea. Genome Biology and Evolution 6, no. 1:192–212. doi: 10.1093/gbe/evu004.
Roberts, E., A. Sethi, J. Montoya, C. R. Woese, and Z. Luthey-Schulten. 2008. Molecular signatures of ribosomal evolution. Proceedings of the National Academy of Sciences U.S.A 105, no. 37:13953–13958. doi: 10.1073/pnas.0804861105.
Rochette, N. C., C. Brochier-Armanet, and M. Gouy. 2014. Phylogenomic test of the hypotheses for the evolutionary origin of eukaryotes. Molecular Biology and Evolution 31, no. 4:832–845. doi: 10.1093/molbev/mst272.
Rosindell, J., and L. J. Harmon. 2012. OneZoom: a fractal explorer for the tree of life. PLoS Biol 10, no. 10:e1001406. doi: 10.1371/journal.pbio.1001406.
Scherl, A., Y. Couté, C. Déon, A. Callé, K. Kindbeiter, J. C. Sanchez, A. Greco, D. Hochstrasser, and J. J. Diaz. 2002. Functional proteomic analysis of human nucleolus. Molecular Biology of the Cell 13, no. 11:4100–4009. doi: 10.1091/mbc.E02-05-0271.
Shajani, Z., M. T. Sykes, and J. R. Williamson. 2011. Assembly of bacterial ribosomes. Annual Review of Biochemistry 80:501–526. doi: 10.1146/annurev-biochem-062608-160432.
Shandilya, J., and S. G. Roberts. 2012. The transcription cycle in eukaryotes: from productive initiation to RNA polymerase II recycling. Biochimica et Biophysica Acta 1819, no. 5:391–400. doi: 10.1016/j.bbagrm.2012.01.010.
Skarstad, K., and T. Katayama. 2013. Regulating DNA replication in bacteria. Cold Spring Harbor Perspectives in Biology 5, no. 4:a012922. doi: 10.1101/cshperspect.a012922.
Sosnowski, B. A., J. M. Belote, and M. McKeown. 1989. Sex-specific alternative splicing of RNA from the transformer gene results from sequence-dependent splice site blockage. Cell 58, no. 3:449–459.
Stepankiw, N., A. Kaidow, E. Boye, and D. Bates. 2009. The right half of the Escherichia coli replication origin is not essential for viability, but facilitates multi-forked replication. Molecular Microbiology 74, no. 2:467–479. doi: 10.1111/j.1365-2958.2009.06877.x.
Strunk, B. S., and K. Karbstein. 2009. Powering through ribosome assembly. RNA 15, no. 12:2083–2104. doi: 10.1261/rna.1792109.
Suárez-Diaz, E., and V. H. Anaya-Muñoz. 2008. History, objectivity, and the construction of molecular phylogenies. Studies in History and Philosophy of Biological and Biomedical Sciences 39, no. 4:451–468. doi: 10.1016/j.shpsc.2008.09.002.
Szafranski, P., Y. Yang, M. U. Nelson, M. J. Bizzarro, R. A. Morotti, C. Langston, and P. Stankiewicz. 2013. Novel FOXF1 deep intronic deletion causes lethal lung developmental disorder, alveolar capillary dysplasia with misalignment of pulmonary veins. Human Mutation 34, no. 11:1467–1471. doi: 10.1002/humu.22395.
Tan, C., and J. Tomkins. 2015. Information processing differences between archaea and eukaraya—Implications for homologs and the myth of eukaryogenesis. Answers Research Journal 8:121–141.
Tang, J. Y., J. C. Lee, M. F. Hou, C. L. Wang, C. C. Chen, H. W. Huang, and H. W. Chang. 2013. Alternative splicing for diseases, cancers, drugs, and databases. Scientific World Journal 2013:703568. doi: 10.1155/2013/703568.
Tiengwe, C., L. Marcello, H. Farr, C. Gadelha, R. Burchmore, J. D. Barry, S. D. Bell, and R. McCulloch. 2012. Identification of ORC1/CDC6-interacting factors in Trypanosoma brucei reveals critical features of origin recognition complex architecture. PLoS One 7, no. 3:e32674. doi: 10.1371/journal.pone.0032674.
Tomkins, J. P., and J. Bergman. 2011. Telomeres: implications for aging and evidence for intelligent design. Journal of Creation 25, no. 1:86–97.
Tomkins, J., and J. Bergman. 2013. Incomplete lineage sorting and other ‘rogue’ data fell the tree of life. Journal of Creation 27, no. 3:84–92.
Tropp, B. E. 2012. Molecular biology: Genes to proteins. 4th ed. Sudbury, Massachusetts: Jones and Bartlett Publishers.
Tucker, C. L., and S. Fields. 2003. Lethal combinations. Nature Genetics 35, no. 3:204–205. doi: 10.1038/ng1103-204.
Tuteja, N., and R. Tuteja. 2004. Prokaryotic and eukaryotic DNA helicases. Essential molecular motor proteins for cellular machinery. European Journal of Biochemistry 271, no. 10:1835–1848. doi: 10.1111/j.1432-1033.2004.04093.x.
Waterhouse, R. M., F. Tegenfeldt, J. Li, E. M. Zdobnov, and E. V. Kriventseva. 2013. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Research 41:D358–365.
Wijaya, E., M. C. Frith, P. Horton, and K. Asai. 2013. Finding protein-coding genes through human polymorphisms. PLoS One 8, no. 1:e54210. doi: 10.1371/journal.pone.0054210.
Wilcox, A. J., C. R. Weinberg, J. F. O’Connor, D. D. Baird, J. P. Schlatterer, R. E. Canfield, E. G. Armstrong, and B. C. Nisula. 1988. Incidence of early loss of pregnancy. New England Journal of Medicine 319, no. 4:189–194. doi: 10.1056/NEJM198807283190401.
Wolf, Y. I., P. S. Novichkov, G. P. Karev, E. V. Koonin and D. J. Lipman. 2009. The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proceedings of the National Academy of Sciences U.S.A 106, no. 18:7273–7280. doi: 10.1073/pnas.0901808106.
Yamamoto, S., V. Bayat, H. J. Bellen, and C. Tan. 2013. Protein phosphatase 1β limits ring canal constriction during Drosophila germline cyst formation. PLoS One 8, no. 7:e70502. doi: 10.1371/journal.pone.0070502.
Yi, F., F. Yang, X. Liu, H. Chen, T. Ji, L. Jiang, X. Wang et al. 2013. RNA-seq identified a super-long intergenic transcript functioning in adipogenesis. RNA Biology 10, no. 6:991–1001. doi: 10.4161/rna.24644.
Zhang, R., and Y. Lin. 2009. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Research 37 (Database issue):D455–458. doi: 10.1093/nar/ gkn858.