The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
An evolutionary argument heavily promoted by the BioLogos organization as evidence of broadscale vertebrate macroevolution is the idea that the human genome contains the 150 base remnant of an egg-yolk related vitellogenin (vtg) gene acquired through descent from a common ancestor shared with chicken. However, research described in this report shows that the alleged vtg fragment in human is not a pseudogene remnant at all, but a functional enhancer element in the fifth intron of a “genomic address messenger” (GAM) gene. This GAM gene produces long noncoding RNAs that have been experimentally shown to selectively inhibit the translation of known target genes, a majority of which are implicated in a variety of human diseases. Messenger RNAs from this gene are also expressed in a variety of human brain tissues. The alleged 150 base vtg sequence contains a variety of highly conserved mammalian transcription factor binding domains, nucleosome depleted open-active chromatin, is hypo-methylated, associates with RNA polymerase 2 in long-range chromatin interactions, and binds the Mafk transcriptional regulator. These combinatorial data clearly show that it is a functional enhancer element in a GAM gene expressed in the human brain—strongly challenging the idea that this sequence is an egg-laying pseudogene genomic fossil.
Keywords: vitellogenin pseudogene, vit1, vtg, BioLogos, evolution
Introduction
Lipoproteins are vital elements in a wide repertoire of biological processes in animals, playing key roles in cell structural support, enzymatic pathways, and cell transport activities. A subset of these lipoproteins are processed in the cell by a diverse set of proteins called large lipid transfer proteins (LLTP). The LLTPs have traditionally been divided up into three general classes 1) vitellogenins (Vtg), 2) the cytosolic large subunit of the microsomal triglyceride transfer protein (MTP), and 3) apolipoproteins (Apo). The apolipoproteins can be further categorized into subgroups which include vertebrate apolipoprotein B (ApoB), crustacean apolipocrustacein (ApoCr), and insect apolipophorin II/I precursor (ApoLp-II/I) (Hayward et al. 2010; Wu, Hui, and Chu 2013).
In non-mammalian vertebrates, the development and establishment of nutritional reserves in the egg yolk are a key factor required for the development of the embryo (Amdam et al. 2003; Byrne, Gruber, and Ab 1989). In living oviparous (egg-laying) terrestrial taxon, especially reptiles and birds, vitellogenins (vtg) play a key role in either transporting or providing the nutritional substrate for proteins, lipids, phosphorous, and calcium in relation to formation of the egg yolk (Romano et al. 2004).
In the grand evolutionary story, it is believed that egg-laying creatures share a common ancestry with placental animals in which the role of vtg proteins were replaced with the development of the placental interface and after birth, continued nutritional sustenance via lactation (Brawand, Wahli, and Kaessmann 2008; Oftedal 2002; Rothchild 2003; Wu, Hui, and Chu 2013). Along these lines, it is believed by some evolutionists that the human genome should contain the remnants of vtg genes—genomic fossils of an ancient egg-laying past. In fact, the BioLogos organization, a religious group of evolutionary scientists and liberal theologians, has been promoting this idea as high level evidence of the grand Darwinian evolutionary hypothesis (Venema 2010, 2012a, b). Interestingly, BioLogos is probably the only evolutionary group that puts such a high level of focus on this hypothesis as key evidence for evolution. Nevertheless, because of the BioLogos outreach and propaganda targeted to the evangelical Christian community to accept hypothetical evolutionary speculation as established scientific fact (Luskin 2014), a more thorough investigation of the vitellogenin pseudogene claims is warranted.
The primary evidence for the BioLogos claims of an ancient egg-laying evolutionary past in the human genome is specifically based on research described in a paper by Brawand, Wahli, and Kaessmann published in 2008. The details of the original claims of sequence similarity by Brawand, Wahli, and Kaessmann are reevaluated using a more recently developed algorithm. But more importantly, the particular region of the genome in question allegedly containing the vtg fragment is more thoroughly investigated in light of seven years of new gene expression and chromatin state research performed by the biomedical genomics community.
Materials and Methods
All genomic sequences were downloaded from the UCSC genome browser website using either the web interface or a Perl script written by author Tomkins. Pairwise DNA alignments were performed using the Geneious software package with the following parameters: global alignment with free end gaps, cost matrix of identity 1.0/0.0, gap open penalty of 3, and a gap extension penalty of 3. These parameters were employed due to the low homology of the sequences being aligned. All gene prediction, ENCODE related data, gene expression data, and other similar biochemical data and annotations were determined using information publicly available in the UCSC genome browser (genome.ucsc.edu).
Results
Sequence similarity
The sequence identified by Brawand, Wahli, and Kaessmann (2008) as being a vtg pseudogene is only 150 bases long and listed in the UCSC genome browser at the genome coordinates of chr1:79254632-79254781 (version hg19). The sequence homology of this 150 base fragment to the alleged ancestral chicken vtg1 gene region is so low that standard DNA alignment algorithms (e.g. BLASTN) are not able to align it. Brawand, Wahli, and Kaessmann first identified the vtg pseudogene fragment with the software WU-BLAST (now deprecated), a tool used to find regions of sequence similarity with minimum loss of sensitivity. Then, they compared the candidate vtg region alignments to those of random genomic background sequence in which it was statistically shown that it was more significantly similar to vtg1 of chicken. They then concluded that it was an authentic vtg gene pseudogene fragment. However, they never definitively reported what the actual level of sequence identity was to chicken vtg1 (exon 3) to which it apparently shared homology.
To ascertain what the real level of sequence identity of the alleged vtg pseudogene fragment was to the chicken vtg1 genomic sequence, both were downloaded from the UCSC genome database. The chicken vtg1 genomic region, not including five prime promoter sequence, is 42,637 bases long (chr8:17537100-17579736) and is a large and complex gene with 35 exons. Thus, a mere 150 base fragment of alleged similarity is but only 0.35% of the actual size of a real functional vtg1 gene in chicken and hardly representative of anything approximating a real pseudogene. Nevertheless, sequence similarity of the alleged vtg fragment compared to the chicken vtg gene was undertaken.
When the human vtg pseudogene fragment was aligned using very liberal gapping parameters (see Materials and Methods) to the chicken genomic sequence, sequence identity was only 62%. Genomic DNA surrounding this fragment was sequentially increased three-fold in size (symmetrically) and each fragment aligned up to 36,450 bases of human genomic DNA. Sequence identity dropped as the fragment size increased, eventually leveling off to about 39% identity for a region of 36,450 bases. This very low level of sequence similarity is most likely due to the large amount of repetitive sequence associated with a broad diversity of somewhat conserved vertebrate transposable elements as shown in Fig. 1A.
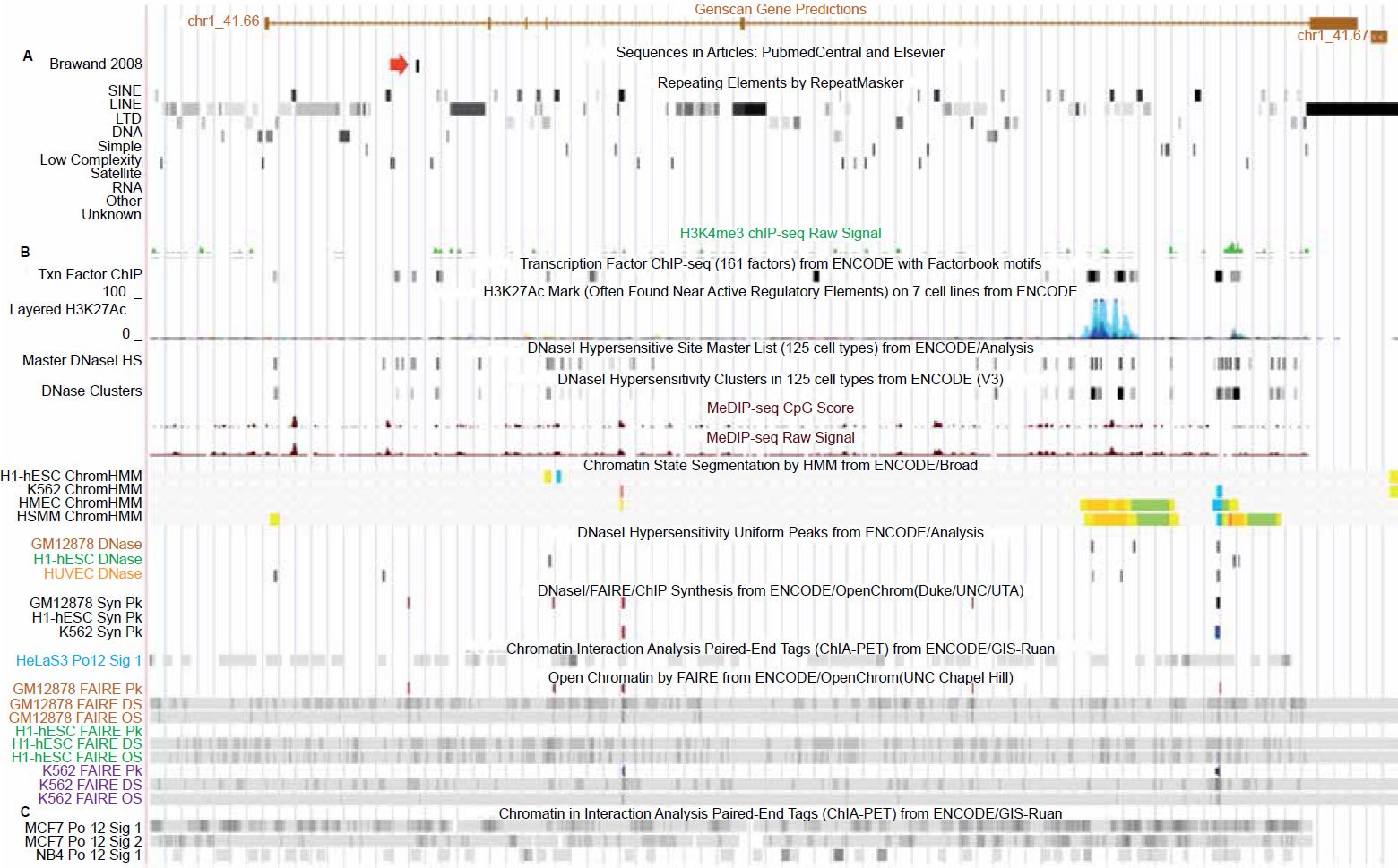
Fig. 1. UCSC genome browser tracks for the GAM gene showing repetitive element content and chromatin state related tracks (alleged vtg fragment marked by red arrow). A. Repetitive element content across the GAM gene. B. Chromatin state tracks showing transcription factor binding, open/active chromatin, transcriptionally active histone modifications, and polymerase2 binding. C. Chromatin interaction analysis by paired-end tags (ChIA-PET) showing that polymerase2 is involved in extensive chromatin looping across the entire gene including the region containing the vtg fragment. View larger image.
The alleged vtg fragment is located inside a functional gene
Outside the questionable sequence identity for this DNA fragment representing a real vtg pseudogene, is the fact that the 150 base sequence in question is located inside a functional gene (figs. 1 and 2). There are multiple lines of combinatorial evidence that indicate this to be the case.
The first line of evidence is that the gene prediction software Genscan (Burge and Karlin 1998) employed on the human genome, as shown in the UCSC genome browser, has predictively annotated a gene of six exons spanning the alleged vtg fragment, which is situated in the last intron (figs. 1 and 2). Bolstering the fact that this is a functional sequence, is gene expression data that runs across the entire length of the predicted gene (fig. 2). More specifically, highly specialized gene expression experiments have functionally identified these expressed sequences in this gene as belonging to a group of novel “genomic address messenger” or “GAM” genes (Bentwich 2007). These GAM genes have been demonstrated to selectively inhibit the translation of known target genes, a majority of which are implicated in a variety of human diseases.
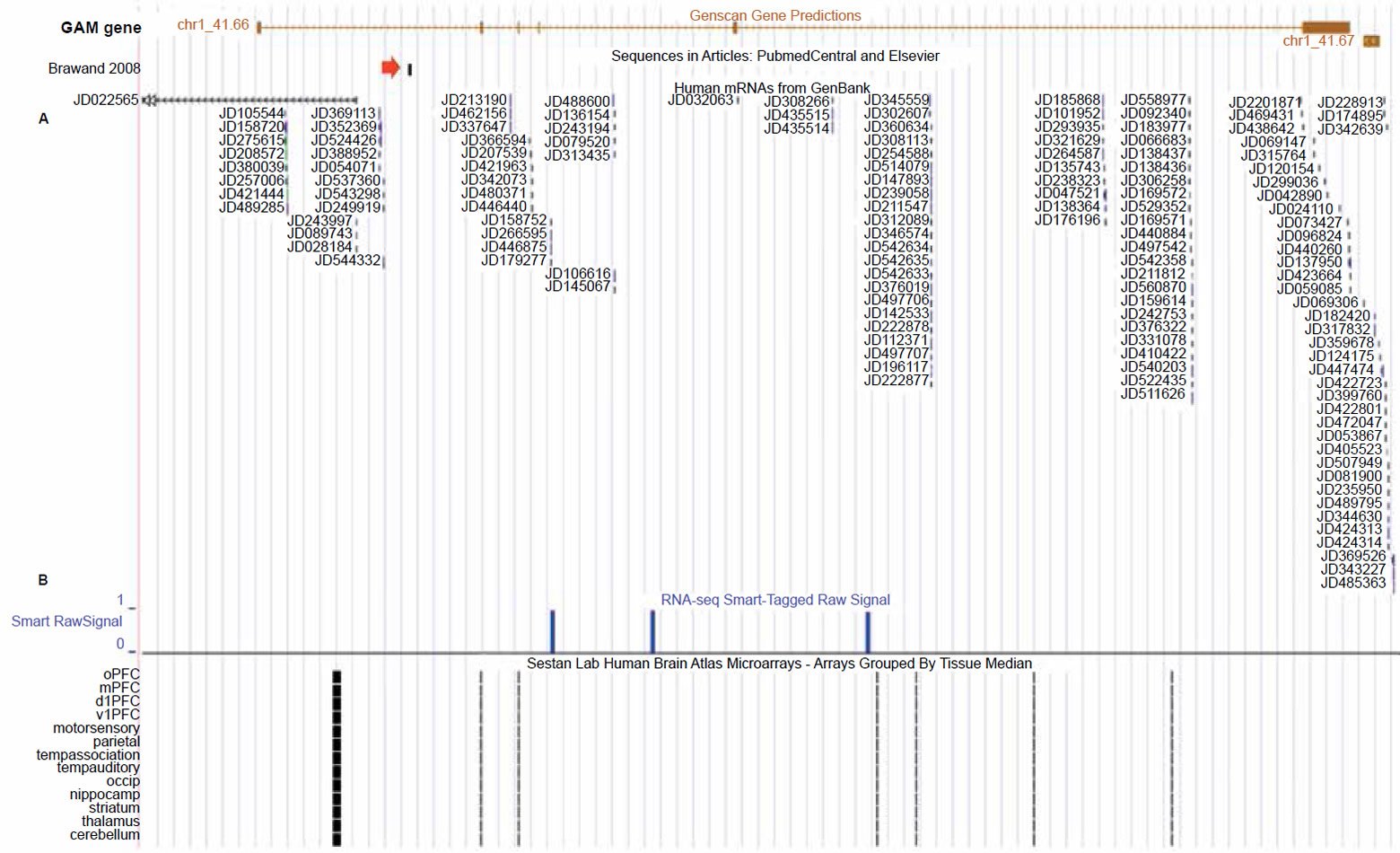
Fig. 2. UCSC genome browser tracks for the GAM gene showing gene expression activity for the GAM gene (alleged vtg fragment marked by red arrow). A. Extensive short DNA sequence reads from specialized gene expression experiments targeting expressed sequences as belonging to a group of “genomic address messenger” or “GAM” genes (Bentwich 2007). B. RNA-seq transcripts generated from the postmortem human frontal cortex gray matter of a 57 year old male (Maunakea et al. 2010). C. Gene expression data from the human brain atlas project using Affymetrix GeneChip microarrays and RNA extracted from both fetal and mature postmortem individuals (Kang et al. 2011; Pletikos et al. 2014). View larger image.
In addition to the GAM gene study, another research project identified expressed sequences from this gene associated with the human brain (the frontal cortex region) based on RNA extracted from a 57 year old male (Maunakea et al. 2010). In confirmation of the RNA sequencing studies for this GAM gene in human brain tissue, the HBT (Human Brain Transcriptome) project at the Department of Neurobiology Yale University School of Medicine also deposited data in the UCSC genome browser from extensive microarray studies showing that this particular GAM gene is not only expressed in the brain tissues of mature humans, but also in the developing fetus (Kang et al. 2011; Pletikos et al. 2014).
Thus, there is strong evidence that this gene produces a type of long noncoding RNA that is involved in post-transcriptional gene regulation in neural tissue. Long noncoding RNAs are proving to be key regulators of nearly all aspects of chromatin function/organization, gene expression, cell signaling, and other cellular processes (Amort et al. 2013; Batista and Chang 2013; Clark et al. 2013; Geisler and Coller 2013; Liebers, Rassoulzadegan, and Lyko 2014; Maass, Luft, and Bahring 2014; Mercer and Mattick 2013; Morris and Mattick 2014; Nie et al. 2012; Rinn and Chang 2012; St Laurent, Savva, and Kapranov 2012; Tomkins 2015; Yoon, Abdelmohsen, and Gorospe 2013).
Further bolstering the fact that a predicted gene with a functionally specified sequence surrounds the alleged vtg fragment, are multiple lines of combinatorial chromatin marks that show it is functionally active (fig. 1). These lines of evidence—including transcriptionally active histone modifications (H3K27Ac, H3K4me3), open and accessible chromatin depleted of nucleosomes, and a wide diversity of transcription factor binding (including RNA polymerase2)— all unequivocal combinatorial chromatin state hallmarks of functional gene sequence (Djebali et al. 2012; Gerstein et al. 2012; Kimura 2013; Li et al. 2012; Neph et al. 2012; Sandstrom et al. 2014; Thurman et al. 2012; Tomkins 2013a, b; Zentner and Henikoff 2013; Zhu et al. 2013).
While much of the epigenetic data for this gene encompassing the alleged vtg fragment was derived from a variety of human cell lines, a significant portion of the data also came from a genomewide epigenetic study of postmortem tissue taken from the frontal cortex gray matter of a 57 year old human male as mentioned above (Maunakea et al. 2010). This diverse data set produced by Maunakea et al. not only included gene expression data from the GAM gene surrounding the alleged vtg fragment but also contained a wide variety of transcriptionally active histone modifications associated with functional gene sequences. So not only is this gene implicated in post-transcriptional gene regulation as a noncoding RNA, but we also have extensive data showing that it is involved in neurological processes in both the embryonic and mature human brain.
The alleged vtg fragment is a transcription factor binding domain
Not only is the alleged vtg fragment positioned inside an expressed long noncoding RNA gene associated with selectively inhibiting the translation of known target genes being expressed in brain tissue, but it also represents a functional sequence associated with transcription factor binding (fig. 3). This contention is based on multiple lines of bioinformatic and biochemical evidence.
The first line of evidence that the alleged vtg fragment contains functional features is that five different highly conserved mammalian transcription factor motifs are identified in the middle of the 150 base sequence for the following factors: FREAC3, FREAC2, FOX04, FOX FOX03, FOX01, and SRY (fig. 3). This particular track in the UCSC genome browser demarcates transcription factor binding sites conserved in the human-mouse-rat alignment where a binding site is determined to be conserved if its score meets the threshold for its binding matrix in all three taxa. This data is largely bioinformatically resolved, but is often corroborated by chromatin marks and other biochemical data (Buck-Koehntop and Defossez 2013; Chen et al. 2015; Choy et al. 2010; Fournier et al. 2012; Neph et al. 2012; Sheffield et al. 2013; Thurman et al. 2012; Whitfield et al. 2012) as is the case for this region of the human genome. In fact, the entire 150 base sequence of the alleged vtg fragment is characterized by a central dip (lowering) in cytosine methylation directly within the predicted binding domains mentioned above, combined with open-active chromatin profiles associated with nucleosome depletion (fig. 3). All of these chromatin state hallmarks are indicative of the predicted binding sites being active and functional.
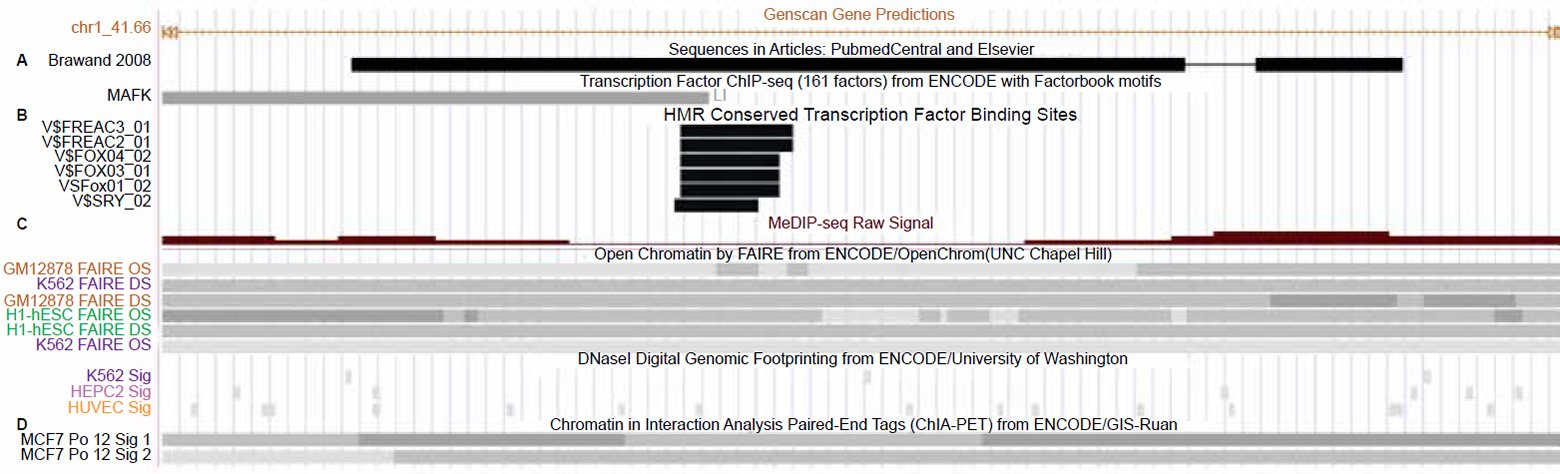
Fig. 3. Close-up view of the alleged 150 base vtg pseudogene fragment in the UCSC genome browser. A. Transcription factor binding track for Mafk overlapping the alleged vtg pseudogne fragment. B. Conserved mammalian transcription factor binding sites in the center of the alleged vtg pseudogene. C. Open-active chromatin and hypomethylation tracks overlapping the transcription factor binding sites. D. Chromatin interaction analysis of paired-end tags (ChIA-PET) tracks showing that this region is involved in long range chromatin interactions associated with RNA polymerase2 and gene transcription. View larger image.
Not only is this alleged vtg regio n replete with transcription factor binding sites and accompanying active-open chromatin marks, but biochemical data for polymerase 2 binding associated with longrange chromatin interactions is also documented in this region along with evidence of binding for the transcription factor MafK (fig. 3). The Maf proteins (MafF, MafG, or MafK) behave as either transcriptional repressors or activators and are key molecular switches functioning in enhancer elements within or outside genes. A classic example is the role of Maf proteins interacting with NFE2 proteins in the locus control region (enhancer-like region) of the beta globin genes (Noordermeer and de Laat 2008; Toki et al. 1997).
When Maf proteins serve as transcriptional activators, they help to recruit other transcription factors to specific DNA-binding sites such as those within this alleged vtg fragment. These data, combined with the location of the alleged vtg region towards the three prime end of this GAM gene suggests a key role for the alleged vtg sequence in long range chromatin interactions associated with both the regulation and transcription of the gene. In fact, chromatin interaction analysis of paired-end tags (ChIA-PET) tracks showing that this region is involved in long range chromatin interactions associated with RNA polymerase2 and gene transcription are shown in Fig. 3.
These enhancer sites within and around active genes are part of the three-dimensional looping of chromatin and the binding of cis-regulatory regions involved with transcription (Dickel, Visel, and Pennacchio 2013; Harmston and Lenhard 2013; Sanyal et al. 2012). In stark contrast to the claims that this 150 base region is nothing but a pseudogene remnant, these data clearly show that instead it is an important enhancer element within a GAM gene expressed in neural tissue.
Evaluating alleged genomic synteny of the vtg fragment
One of the supporting arguments for the vtg pseudogene fragment being authentic is that it shares gene neighborhood synteny with chicken. When this was investigated, it was found that gene synteny surrounding the chicken vtg1 gene (~360,000 bases) compared to the region surrounding the alleged vit1 fragment in human, was completely different except for the presence of the LTD1 gene which was about three times the distance (~100,000 bases) from the alleged vit fragment as its homolog is in chicken (~38,000 bases). Most importantly, the functional chicken vtg1 gene, covers over 42,000 bases of genomic sequence. However, the fact that the alleged vtg fragment is a functional enhancer element inside an active GAM gene completely unrelated to vitellogenin protein production renders the argument of synteny null and void to begin with.
Summary
The whole evolutionary argument for pseudogenes being nothing but dead genomic fossils is now coming under serious scrutiny by both creationists and evolutionists alike (Ala et al. 2013; Bergman 2013; Taulli, Loretelli, and Pandolfi 2013; Tomkins 2013b, 2014; Wen et al. 2012). As genomics research progresses, it is becoming clear that many pseudogenes produce regulatory noncoding RNAs that are key players in human health and disease. In fact, in a recent report defining ENCODE data related to pseudogenes, only 20% of the annotated pseudogenes in humans lack any signs of experimentally defined biochemical activity (Sisu et al. 2014). As ENCODE research progresses, this number will probably continue to decrease. Nevertheless, the alleged vtg pseudogene fragment in human does not really qualify as a pseudogene since it is such an incredibly small and dissimilar feature compared to the supposed ancestral sequence from which it allegedly was derived.
The BioLogos organization promotes hypothetical broad-scale vertebrate macroevolution as real science (Luskin 2014). One of the chief arguments they put forth as evolutionary proof is the idea that the human genome contains the 150 base remnant of an eggyolk related vitellogenin (vtg) gene acquired through descent from a common ancestor shared with chicken. However, research described in this report shows that the alleged vtg fragment in human is only 62% identical to its alignable counterpart in the chicken vtg1 gene (exon 3). Moreover, the actual chicken vtg1 gene is 42,637 bases long (not including promoter sequence) so the alleged vtg fragment in human actually represents less than 1% of the original ancestral gene. Even in an evolutionary sense, to say that a pseudogene can be identified by only 0.35% of the original sequence is quite a stretch of the Darwinian paradigm.
However, the real story is that the alleged 150 base vtg sequence is not a pseudogene remnant at all, but a functional enhancer element in the fifth intron of a “genomic address messenger” (GAM) gene. This particular GAM gene produces long noncoding RNAs that have been experimentally shown to selectively inhibit the translation of known target genes, a majority of which have been implicated in a variety of human diseases. Messenger RNAs from this particular gene are also known to be expressed in a variety of human brain tissues in both fetal and mature subjects in three separate studies.
All of the combinatorial data presented in this report clearly show that the alleged vtg pseudogene fragment is a functional enhancer element in a GAM gene expressed in the human brain—overturning the idea that this sequence is an egg-laying pseudogene genomic fossil.
References
Ala, U., F. A. Karreth, C. Bosia, A. Pagnani, R. Taulli, V. Léopold, Y. Tay, P. Provero, R. Zecchina, and P. P. Pandolfi. 2013. “Integrated Transcriptional and Competitive Endogenous RNA Networks are Cross-Regulated in Permissive Molecular Environments.” Proceedings of the National Academy of Sciences USA 110 (18): 7154–59. doi:10.1073/pnas.1222509110.
Amdam, G. V., K. Norberg, A. Hagen, and S. W. Omholt. 2003. “Social Exploitation of Vitellogenin.” Proceedings of the National Academy of Sciences USA 100 (4): 1799–802. doi:10.1073/pnas.0333979100.
Amort, T., M. F. Soulière, A. Wille, X-Y. Jia, H. Fiegl, H. Wörle, R. Micura, and A. Lusser. 2013. “Long Non-Coding RNAs as Targets for Cytosine Methylation.” RNA Biology 10 (6): 1003–08. doi:10.4161/rna.24454.
Batista, P. J., and H. Y. Chang. 2013. “Long Noncoding RNAs: Cellular Address Codes in Development and Disease.” Cell 152 (6): 1298–307. doi:10.1016/j.cell.2013.02.012.
Bentwich, I. 2007. “Bioinformatically Detectable Group of Novel Regulatory Genes and Uses Thereof.” In United States Patent: Rosetta Genomics Ltd. http://www.google. com/patents/US7250496.
Bergman, J. 2013. “The Origins and Genetic Functions of Pseudogenes.” Creation Research Society Quarterly 49: 308–18.
Brawand, D., W. Wahli, and H. Kaessmann. 2008. “Loss of Egg Yolk Genes in Mammals and the Origin of Lactation and Placentation.” PLoS Biol 6 (3): e63. doi:10.1371/journal. pbio.0060063.
Buck-Koehntop, B. A., and P. A. Defossez. 2013. “On How Mammalian Transcription Factors Recognize Methylated DNA.” Epigenetics 8 (2): 131–37. doi:10.4161/epi.23632.
Burge, C. B., and S. Karlin. 1998. “Finding the Genes in Genomic DNA.” Current Opinion in Structural Biology 8 (3): 346–54.
Byrne, B. M., M. Gruber, and G. Ab. 1989. “The Evolution of Egg Yolk Proteins.” Progress in Biophysics and Molecular Biology 53 (1): 33–69.
Chen, H., J. Chen, L. A. Muir, S. Ronquist, W. Meixner, M. Ljungman, T. Ried, S. Smale, and I. Rajapakse. 2015. “Functional Organization of the Human 4D Nucleome.” Proceedings of the National Academy of Sciences USA 112 (26): 8002–07. doi:10.1073/pnas.1505822112.
Choy, M-K., M. Movassagh, H-G. Goh, M. R. Bennett, T. A. Down, and R. S-Y. Foo. 2010. “Genome-Wide Conserved Consensus Transcription Factor Binding Motifs are Hyper-Methylated.” BMC Genomics 11:519. doi:10.1186/1471-2164-11-519.
Clark, M. B., A. Choudhary, M. A. Smith, R. J. Taft, and J. S. Mattick. 2013. “The Dark Matter Rises: The Expanding World of Regulatory RNAs.” Essays in Biochemistry 54: 1–16. doi:10.1042/bse0540001.
Dickel, D. E., A. Visel, and L. A. Pennacchio. 2013. “Functional Anatomy of Distant-Acting Mammalian Enhancers.” Philosophical Transactions of the Royal Society of London B Biological Sciences 368 (1620): 20120359. doi:10.1098/rstb.2012.0359.
Djebali, S., C. A. Davis, A. Merkel, A. Dobin, T. Lassmann, A. Mortazavi, A. Tanzer, et al. 2012. “Landscape of Transcription in Human Cells.” Nature 489 (7414): 101–08. doi:10.1038/nature11233.
Fournier, A., N. Sasai, M. Nakao, and P. A. Defossez. 2012. “The Role of Methyl-Binding Proteins in Chromatin Organization and Epigenome Maintenance.” Briefings in Functional Genomics 11 (3): 251–64. doi:10.1093/bfgp/elr040.
Geisler, S., and J. Coller. 2013. “RNA in Unexpected Places: Long Non-Coding RNA Functions in Diverse Cellular Contexts.” National Review of Molecular Cell Biology 14 (11): 699–712. doi:10.1038/nrm3679.
Gerstein, M. B., A. Kundaje, M. Hariharan, S. G. Landt, K. K. Yan, C. Cheng, X. J. Mu, et al. 2012. “Architecture of the Human Regulatory Network Derived from ENCODE Data.” Nature 489 (7414): 91–100. doi:10.1038/nature11245.
Harmston, N., and B. Lenhard. 2013. “Chromatin and Epigenetic Features of Long-Range Gene Regulation.” Nucleic Acids Research 41 (15): 7185–99. doi:10.1093/nar/gkt499.
Hayward, A., T. Takahashi, W. G. Bendena, S. S. Tobe, and J. H. Hui. 2010. “Comparative Genomic and Phylogenetic Analysis of Comparative Genomic and Phylogenetic Analysis of Vitellogenin and Other Large Lipid Transfer Proteins in Metazoans. FEBS Letters 584 (6): 1273–78. doi:10.1016/j.febslet.2010.02.056.
Kang, H. J., Y. I. Kawasawa, F. Cheng, Y. Zhu, X. Xu, M. Li, A. M. M. Sousa, et al. 2011. “Spatio-Temporal Transcriptome of the Human Brain.” Nature 478 (7370): 483–89. doi:10.1038/nature10523.
Kimura, H. 2013. “Histone Modifications for Human Epigenome Analysis.” Journal of Human Genetica 58 (7): 439–45. doi:10.1038/jhg.2013.66.
Li, G., X. Ruan, R. K. Auerbach, K. S. Sandhu, M. Zheng, P. Wang, H. Mei, et al. 2012. “Extensive Promoter-Centered Chromatin Interactions Provide a Topological Basis for Transcription Regulation.” Cell 148 (1–2): 84–98. doi:10.1016/j.cell.2011.12.014.
Liebers, R., M. Rassoulzadegan, and F. Lyko. 2014. “Epigenetic Regulation by Heritable RNA.” PLoS Genetics 10 (4): e1004296. doi:10.1371/journal.pgen.1004296.
Luskin, C. 2014. “The New Theistic Evolutionists: BioLogos and the Rush to Embrace the ‘Consensus’.” Christian Research Journal 37 (3): 32–41.
Maass, P. G., F. C. Luft, and S. Bähring. 2014. “Long Non-Coding RNA in Health and Disease.” Journal of Molecular Medicine (Berlin, Germany) 92 (4): 337–46. doi:10.1007/s00109-014-1131-8.
Maunakea, A. K., R. P. Nagarajan, M. Bilenky, T. J. Ballinger, C. D’Souza, S. D. Fouse, B. E. Johnson, et al. 2010. “Conserved Role of Intragenic DNA Methylation in Regulating Alternative Promoters.” Nature 466 (7303): 253–57. doi:10.1038/nature09165.
Mercer, T. R., and J. S. Mattick. 2013. “Structure and Function of Long Noncoding RNAs in Epigenetic Regulation.” Nature Structural and Molecular Biology 20 (3): 300–07. doi:10.1038/nsmb.2480.
Morris, K. V., and J. S. Mattick. 2014. “The Rise of Regulatory RNA.” Nature Reviews Genetics 15 (6): 423–37. doi:10.1038/nrg3722.
Neph, S., J. Vierstra, A. B. Stergachis, A. P. Reynolds, E. Haugen, B. Vernot, R. E. Thurman, et al. 2012. “An Expansive Human Regulatory Lexicon Encoded in Transcription Factor Footprints.” Nature 489 (7414): 83–90. doi:10.1038/nature11212.
Nie, L., H-J. Wu, J-M. Hsu, S-S. Chang, A. M. LaBaff, C-W. Li, Y. Wang, J. L. Hsu, and M-C. Hung. 2012. “Long Non-Coding RNAs: Versatile Master Regulators of Gene Expression and Crucial Players in Cancer.” American Journal of Translational Research 4 (2): 127–50.
Noordermeer, D., and W. de Laat. 2008. “Joining the Loops: Beta-Globin Gene Regulation.” IUBMB Life 60 (12): 824–33. doi:10.1002/iub.129.
Oftedal, O. T. 2002. “The Mammary Gland and Its Origin During Synapsid Evolution.” Journal of Mammary Gland Biology and Neoplasia 7 (3): 225–52.
Pletikos, M., A. M. M. Sousa, G. Sedmak, K. A. Meyer, Y. Zhu, F. Cheng, M. Li, Y. I. Kawasawa, and N. Šestan. 2014. “Temporal Specification and Bilaterality of Human Neocortical Topographic Gene Expression.” Neuron 81 (2): 321–32. doi:10.1016/j.neuron.2013.11.018.
Rinn, J. L., and H. Y. Chang. 2012. “Genome Regulation by Long Noncoding RNAs.” Annual Reviews of Biochemistry 81: 145–66. doi:10.1146/annurevbiochem-051410-092902.
Romano, M., P. Rosanova, C. Anteo, and E. Limatola. 2004. “Vertebrate Yolk Proteins: A Review.” Molecular Reproduction and Development 69 (1): 109–16. doi:10.1002/mrd.20146.
Rothchild, I. 2003. “The Yolkless Egg and the Evolution of Eutherian Viviparity.” Biology of Reproduction 68 (2): 337–57.
Sandstrom, R. S., M. R. Foret, D. A. Grow, E. Haugen, C. T. Rhodes, A. E. Cardona, C. F. Phelix, Y. Wang, M. S. Berger, and C. H. Lin. 2014. “Epigenetic Regulation by Chromatin Activation Mark H3K4me3 in Primate Progenitor Cells within Adult Neurogenic Niche.” Scientific Reports 4: 5371. doi:10.1038/srep05371.
Sanyal, A., B. R. Lajoie, G. Jain, and J. Dekker. 2012. “The Long-Range Interaction Landscape of Gene Promoters.” Nature 489 (7414): 109–13. doi:10.1038/nature11279.
Sheffield, N. C., R. E. Thurman, L. Song, A. Safi, J. A. Stamatoyannopoulos, B. Lenhard, G. E. Crawford, and T. S. Furey. 2013. “Patterns of Regulatory Activity Across Diverse Human Cell Types Predict Tissue Identity, Transcription Factor Binding, and Long-Range Interactions.” Genome Research 23 (5): 777–88. doi:10.1101/gr.152140.112.
Sisu, C., B. Pei, J. Leng, A. Frankish, Y. Zhang, S. Balasubramanian, R. Harte, et al. 2014. “Comparative Analysis of Pseudogenes Across Three Phyla.” Proceedings of the National Academy of Sciences USA 111 (37): 13361–66. doi:10.1073/pnas.1407293111.
St Laurent, G., Y. A. Savva, and P. Kapranov. 2012. “Dark Matter RNA: An Intelligent Scaffold for the Dynamic Regulation of the Nuclear Information Landscape.” Frontiers in Genetics 3: 57. doi:10.3389/fgene.2012.00057.
Taulli, R., C. Loretelli, and P. P. Pandolfi. 2013. “From Pseudo-ceRNAs to circ-ceRNAs: A Tale of Cross-Talk and Competition.” Nature Structural and Molecular Biology 20 (5): 541–43. doi:10.1038/nsmb.2580.
Thurman, R. E., E. Rynes, R. Humbert, J. Vierstra, M. T. Maurano, E. Haugen, N. C. Sheffield, et al. 2012. “The Accessible Chromatin Landscape of the Human Genome.” Nature 489 (7414): 75–82. doi:10.1038/nature11232.
Toki, T., J. Itoh, J. Kitazawa, K. Arai, K. Hatakeyama, J. Akasaka, K. Igarashi, et al. 1997. “Human Small Maf Proteins Form Heterodimers with CNC Family Transcription Factors and Recognize the NF-E2 Motif.” Oncogene 14 (16): 1901–10. doi:10.1038/sj.onc.1201024.
Tomkins, J. 2013a. “Alleged Human Chromosome 2 ‘Fusion Site’ Encodes an Active DNA Binding Domain Inside a Complex and Highly Expressed Gene—Negating Fusion.” Answers Research Journal 6: 367–75.
Tomkins, J.P. 2013b. “The Human Beta-Globin Pseudogene is Non-Variable and Functional.” Answers Research Journal 6: 293–301.
Tomkins, J. 2014. “The Human GULO Pseudogene—Evidence for Evolutionary Discontinuity and Genetic Entropy.” Answers Research Journal 7: 91–101.
Tomkins, J. 2015. “Extreme Information: Biocomplexity of Interlocking Genome Languages.” Creation Research Society Quarterly 51 (3): 187–201.
Venema, D. 2010. “Signature in the Pseudogenes, Part 2.” The BioLogos Forum. http://old.biologos.org/blog/series/signature-in-the-pseudogenes.
Venema, D. 2012a. “ENCODE and ‘Junk DNA,’ Part 2: Function: What’s in a Word?” http://biologos.org/blog/encode-and-junk-dna-part-2.
Venema, D. 2012b. “Is There ‘Junk’ in Your Genome? Part 4.” http://biologos.org/blog/understanding-evolution-is-therejunk-in-your-genome-part-4.
Wen, Y. Z., L. L. Zheng, L. H. Qu, F. J. Ayala, and Z. R. Lun. 2012. “Pseudogenes Are Not Pseudo Any More.” RNA Biology 9 (1): 27–32. doi:10.4161/rna.9.1.18277.