The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
Modern genomics provides the ability to screen the DNA of a wide variety of organisms to scrutinize broken metabolic pathways. This wealth of data has revealed wide-spread genetic entropy in human and other genomes. Loss of the vitamin C pathway due to deletions in the GULO (L-gulonolactone oxidase) gene has been detected in humans, apes, guinea pigs, bats, mice, rats, pigs, and passerine birds. Contrary to the popularized claims of some evolutionists and neo-creationists, patterns of GULO degradation are taxonomically restricted and fail to support macroevolution. Current research and data reported here show that multiple GULO exon losses in human, chimpanzee, and gorilla occurred independently in each taxon and are associated with regions containing a wide variety of transposable element fragments. Thus, they are another example of sequence deletions occurring via unequal recombination associated with transposable element repeats. The 28,800 base human GULO region is only 84% and 87% identical compared to chimpanzee and gorilla, respectively. The 13,000 bases preceding the human GULO gene, which corresponds to the putative area of loss for at least two major exons, is only 68% and 73% identical to chimpanzee and gorilla, respectively. These DNA similarities are inconsistent with predictions of the common ancestry paradigm. Further, gorilla is considerably more similar to human in this region than chimpanzee—negating the inferred order of phylogeny. Taxonomically restricted gene degradation events are emerging as a common theme associated with genetic entropy and systematic discontinuity, not macroevolution.
Keywords: GULO pseudogene, Human-ape DNA similarity, Comparative genomics, Vitamin C
Introduction
Vitamins are organic compounds which organisms must acquire from their environment because they cannot synthesize them, at least not at levels sufficient to maintain their health. They serve as precursors to cofactors that function with enzymes in both central and secondary metabolic pathways. In fact, the first such compound discovered was labeled a “vital amine” (where we get the term “vitamin”) that was isolated from rice husks (thiamine/Vitamin B1) and prevented the disease beri-beri (Funk 1912). An organism that requires dietary vitamins is referred to as a vitamin “auxotroph”. Patterns of vitamin auxotrophy across the schema of life are complicated and often involve complex patterns of organismal interdependence (Helliwell, Wheeler, and Smith 2013).
An emerging theme from the continuing progression of genomics research across the spectrum of eukaryotic life is the widespread decay of pathways for vitamin-synthesis (Helliwell, Wheeler, and Smith 2013). This paradigm is of great importance to the creationist model of genetic entropy which postulates that genomes are in a continual state of degradation over time, not forward progressing evolution (Sanford 2010). Indeed, several recent reports have shown that a considerable amount of recently acquired variation in the human exome (protein coding regions) the past 5000 to 10,000 years is associated with heritable disease (Fu et al. 2013; Tennessen et al. 2012). Another important component of the creationist model of origins is the idea of molecular discontinuity between unrelated taxon (Tomkins and Bergman 2013). As will be demonstrated in this report, the enigma of the GULO pseudogene analyzed in the light of new genomic evidence most closely aligns with a creationist model incorporating both of these paradigms.
Degradation of the vitamin C pathway is an enigmatic case because many vertebrate species are able to synthesize this vitamin (Drouin, Godin, and Page 2011; Yang 2013). The anti-oxidant vitamin C is a water-soluble molecule that protects organisms from oxidative stress (Nowak, Carter, and Schnellmann 2000) and is essential to collagen production. Production of collagen is necessary to prevent development of the pathology known as scurvy (Gropper and Smith 2012). Other documented functions of vitamin C include its participation in the synthesis of carnitine, neurotransmitters, and the catabolism of tyrosine (Combs 2008; Gropper and Smith 2012; Padh 1990; Pohanka et al. 2012).
Biosynthesis of vitamin C is a multi-step process that starts in mammals with glucose as an initial substrate (Linster and Van Schaftingen 2007). The terminal enzyme in the pathway is L-gulono-γ-lactone oxidase which catalyzes the final step (fig. 1). The gene that encodes this enzyme is typically symbolized in the literature as GULO, GLO, or Gulo—the gene that is the focus of this report.
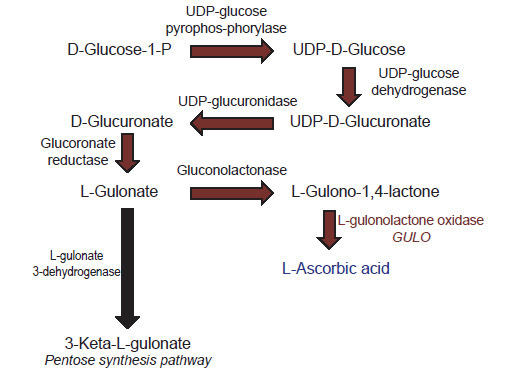
Fig. 1. Depiction of the biochemical pathway for vitamin C biosynthesis in vertebrates.
Loss of GULO gene function primarily impacts the cells ability to make vitamin C (Linster and Van Schaftingen 2007). In contrast, losing the function of genes for other enzymes in the vitamin C pathway directly affects the production of many important biomolecules in the cell because they are multifunctional (Linster and Van Schaftingen 2007). For example, losing gene function for gluconolactonase not only affects the synthesis of L-gulono-1,4-lactone, but also affects caprolactam degradation, the pentose phosphate pathway, and other processes (BRENDA database, http://www.brenda-enzymes.org). Thus, the standard belief among scientists is that the GULO gene may be “predisposed” to being lost or mutated (pseudogenization) compared to other genes because it makes an enzyme that is allegedly unnecessary for other biochemical pathways.
Although not life threatening when supplemented by external sources of vitamin C, the effects from loss of GULO function may be more complicated and pervasive in the cell than previously thought. There is recent evidence from research using a naturally occurring mouse GULO deletion mutant showing that multiple categories of gene expression in the nucleus are markedly affected by GULO inactivation (Jiao et al. 2011; Mohan et al. 2005). Thus, the deactivation of the GULO gene may be more closely related to the idea of cellular network optimization and not what is minimally required for the biochemical survival of the organism. This idea will be revisited later in this report in the summary and discussion.
Evolutionary Anomalies of GULO Gene Loss and Degradation
The macroevolutionary enigmas and contradictions related to vitamin C biosynthesis exist at a variety of levels in the broad schema of life. The first is the fact that yeasts, plants, and animals all use different pathways to synthesize it, so there is no clear ancestral pattern of origin for it in the alleged tree of life and it has arisen suddenly in major taxonomic groups. The yeasts produce a vitamin C variant called D-erythroascorbate from D-arabinose, plants synthesize vitamin C from GDP-D-mannose, and vertebrate animals make vitamin C from the final pathway substrate UDP-D-glucuronate (Hancock, Galpin, and Viola 2000; Linster et al. 2007; Linster and Van Schaftingen 2007; Smirnoff 2001). In addition, it was recently found that GULO genes exist in the urochordate sea squirt and achordate sponge (Yang 2013). However, these invertebrates both possess GULO genes distinctly different in structure from those in vertebrates. It is unknown at this time what the exact nature of the vitamin C pathway is in these organisms.
Another evolutionary anomaly is that the main organ in which vitamin C is synthesized supposedly changed twice from the kidney to the liver during vertebrate macroevolution—at least once in birds and once in mammals (Birney, Jenness, and Hume 1980; Chatterjee 1973; Drouin, Godin, and Page 2011). Vitamin C is produced in the kidneys of non-teleost fishes, amphibians, and reptiles, but in the livers of mammals. And among bird taxa, it is a complete mixture and long-standing evolutionary enigma. Vitamin C is produced solely in the kidneys of some birds, just the livers in others, both livers and kidneys in others, and in a variety of taxon there is no detectable synthesis (Chaudhuri and Chatterjee 1969; Drouin, Godin, and Page 2011; Martinez del Rio 1997; Roy and Guha 1958).
Other anomalies are found when examining the presence and absence of vitamin C synthesis from a broad evolutionary perspective. Of particular note is the growing data set showing that cartilaginous and non-teleost bony fish species are able to synthesize vitamin C while no known teleost (ray-finned) fish has this capability, which is associated with the complete absence of the GULO gene (Drouin, Godin, and Page 2011; Wong et al. 2013; Yang 2013). The complete lack of GULO genes in all studied teleost fish genomes thus far is highly significant because they make up about 95% of all fish taxon (Yang et al. 2013). The key question from a grand evolutionary perspective is how did these genes completely disappear during macroevolution in teleost fishes and then reappear in other metazoan lineages later on? Another interesting anomaly is that the various genes that typically flank GULO in mammal and bird genomes are dispersed throughout the genomes in teleost fishes in variable taxonomically restricted patterns (Yang 2013). Despite their complete lack of a GULO gene, teleost fish successfully obtain their vitamin C requirements from their normal dietary intake in the wild.
The loss of vitamin C synthesis in various passerine birds is due to instances of taxonomically independent deletion mutations in the GULO gene (Drouin, Godin, and Page 2011). Because these GULO mutations in birds do not follow the inferred evolutionary pattern of bird evolution, they are said to be recent events in terminal lineages. However, at least 11 different passerine bird genera with intact functional GULO genes have postulated lineages supposedly derived from ancestors with broken GULO genes—contradicting the standard dogma for bird evolution (Drouin, Godin, and Page 2011).
The initial assessment of bats suggested they lacked the ability to synthesize vitamin C (Birney, Jenness, and Ayaz 1976). However, several recent studies have shown that approximately two thirds of different bat species have functionally expressed GULO genes (Cui et al. 2011a; Cui et al. 2011b). The various bat taxon taxa that lack the ability to make vitamin C have varying levels of deletion degradation in their GULO genes and the patterns of sequence variation show that they are lineage independent events. Trying to explain the discontinuous GULO deletion patterns within a common descent paradigm has produced a variety of difficult contradictions for a coherent model of bat evolution (Cui et al. 2011b).
Loss of vitamin C pathway function and GULO gene degradation has also been detected in guinea pigs, great apes, and humans (Lachapelle and Drouin 2011; Nishikimi, Kawai, and Yagi 1992; Nishikimi et al. 1994; Ohta and Nishikimi 1999). In addition, naturally occurring scurvy and osteogenic disease pathologies related to GULO inactivating mutations and large-scale deletions have been documented in rats, mice, and pigs (Harris et al. 2005; Hasan et al. 2004; Jiao et al. 2005; Kawai et al. 1992; Mohan et al. 2005). Thus, degradation of the GULO gene in a wide array of mammals and birds is a relatively common occurrence.
The GULO gene lies within a gene-dense region in all vertebrate genomes studied thus far (Yang 2013). Related to this fact is the evolutionary anomaly that the gene neighborhood surrounding the GULO locus is rearranged across the vertebrate spectrum of life, and the patterns cannot be readily resolved into the standard inferred evolutionary lineages (Yang 2013). Another interesting discovery related to this issue is the fact that the genes surrounding GULO are each members of gene families with alleged copies located in other regions of the host genome, yet GULO is a unitary gene—not a member of a gene family (Yang 2013). In the standard evolutionary model of gene origins, the majority of new genes are believed to have arisen via the duplication of existing sequences, even though their individual functions may be completely different (Assis and Bachtrog 2013; Innan and Kondrashov 2010). In regard to the GULO locus, the evolutionary mystery recently noted in a study by Yang (2013), is why the GULO gene was not also duplicated along with all of its genomic neighbors.
Human GULO is Degenerate and Chromatin Repressed
In humans, the GULO gene locus appears to be a broken gene suffering major deletions of key exons associated with a functional mammalian GULO gene (fig. 2A). Over the past 25+ years of messenger RNA sequencing from a large diversity of human tissues and cell lines, no GULO transcripts have been detected (Yang 2013). To confirm this status, all six exon sequences in the human GULO pseudogene obtained from the Ensembl database (ensembl.org), were queried against the current nucleotide and EST collections at NCBI and no hits for expressed human GULO sequences were obtained.
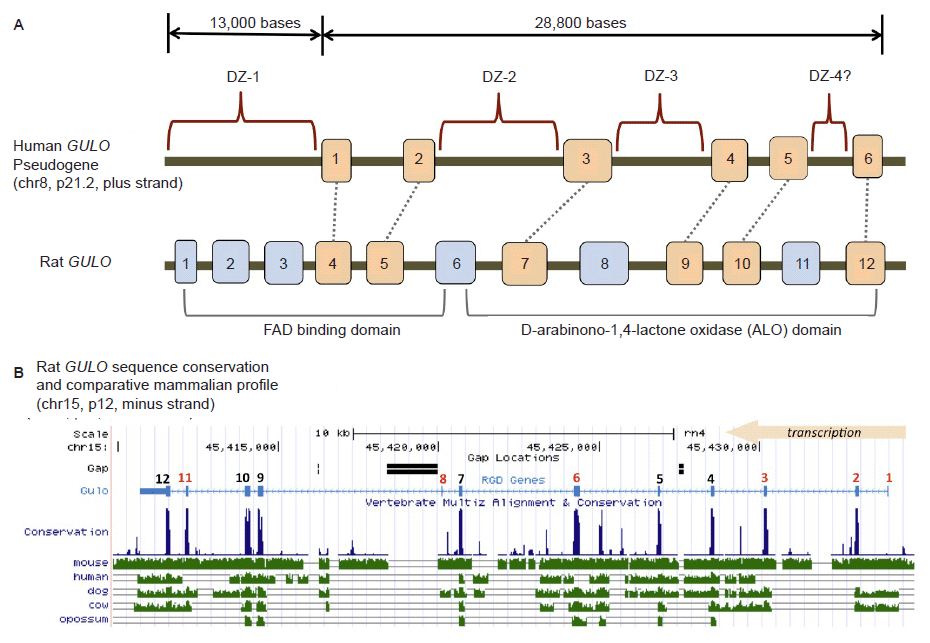
Fig 2. Structure of the human GULO pseudogene. (A) Comparison of the human GULO pseudogene region to the rat functional GULO sequence (adapted in part from data in Yang (2013) and the UCSC genome browser). Exon 1 of the rat GULO gene is missing in other non-rodent mammals. (B) Comparison of several vertebrate GULO gene tracks to the functional rat GULO sequence showing the highly conserved nature of the exons (data extracted from the UCSC genome browser).
Traditionally, the human GULO pseudogene has been compared to the functional rat GULO gene (Nishikimi, Kawai, and Yagi 1992; Nishikimi et al. 1994; Ohta and Nishikimi 1999). According to the UCSC genome browser (genome.ucsc.edu) and the Rat Genome Database (rgd.mcw.edu), the rat GULO gene (chr15, region p12) is oriented and transcribed on the minus strand. Interestingly, the human and ape GULO pseudogenes are oriented in the plus strand configuration (chr8, region p21.2 in human). While the rat GULO gene may serve as a general guide to exon presence and absence in degraded GULO genes in other mammals, the rat GULO is clearly in a different chromosomal configuration (compared to humans and apes) and represents a unique design pattern specific to rodents (mouse GULO is on chr15, minus strand).
When the six human GULO exons were each individually blasted against the nucleotide database at NCBI, the top hit to a functional GULO gene for all six exons was always a mammalian species other than rat (Table 1). In addition, human exons 2 and 6, were only partially similar to rat, and overall only 24.9 and 20.2% similar (respectively) to their corresponding rat exons (Table 1). Thus, the comparative use of the rat GULO gene with human has obvious limitations.
Table 1. The DNA sequence identity for each human GULO exon compared to the top hit to a functional GULO sequence in the NCBI nucleotide database. The rat GULO identities for each exon are also shown. Identities include both insertions and deletions.
| Human GULO exon | Length (bases) | Creature with Highest Functional Sequence Identity | Identity for prior col. | Rat GULO exon identity |
| 1 | 101 | Northern Greater Galago | 87.4% | 85.4% |
| 2 | 96 | Greater False Vampire Bat | 68.1% | 24.9% |
| 3 | 107 | Pratt’s Roundleaf Bat | 80.7% | 71.6% |
| 4 | 151 | Chinese Tree Shrew | 84.1% | 79.5% |
| 5 | 164 | African Bush Elephant | 87.5% | 83.5% |
| 6 | 129 | White Rhinoceros | 87.6% | 20.2% |
Interestingly, it is an alleged single base insertion deletion in the rat GULO DNA sequence in exon ten (compared to humans and apes) that is used as the primary argument for inherited mistakes in human evolution. The errant nature of this evolutionary argument was previously addressed by Truman and Borger (2007) who questioned the validity of using the rat GULO gene as a standard for comparison. In fact, when comparing the complete 164 base sequence of human exon 5 that corresponds to rat exon 10, the overall similarity is only 83.5% (Table 1). Once again, given the marked sequence difference, the question of using rat as a definitive standard becomes questionable. Furthermore, the striking evolutionary discontinuity between humans and apes outside this alleged single base anomaly (based on the rat GULO) documented in this report (below) nullifies the simplistic grossly oversimplified evolutionary argument that this pseudogene supports macroevolution.
When the functional rat GULO gene is broadly compared across diverse metazoan taxon, the exon regions of the gene (except for rat exon 1), are generally well conserved (fig. 2B). Thus, it is reasonable to assume that the presence of similar GULO exon sequences in taxa with broken GULO genes and defunct vitamin C pathways, represent degenerate gene sequences. A consensus functional GULO gene in mammals encodes two basic functional protein domains as shown in fig. 2A (Yang 2013). The putative loss of key exon encoding segments from both these functional regions in the human GULO is also illustrated in fig. 2A.
Human ENCODE information mined from the UCSC genome browser reveals the presence of epigenetic damage control systems in place to silence activity at the alleged degenerate GULO locus and insulate it from neighboring genes. Chromatin state mapping shows that the region is polycomb-repressed (discussed below) and transcriptionally inactive—corresponding with a complete absence of expressed human GULO transcripts. Low activity chromatin state signals, typically associated with heterochromatic regions of the genome, also characterizes the human GULO pseudogene. Also, flanking the GULO pseudogene are regions of chromatin state insulator demarcation that overlap binding sites for the CTCF protein. The multifunctional CTCF protein is implicated in diverse regulatory functions, which includes transcriptional activation and repression, chromatin insulation, genomic imprinting, and spatial organization in the genome (Kapranov and St Laurent 2012; Phillips and Corces 2009). Based on current three-dimensional models of how this concept works, the CTCF sites flanking the degenerate GULO locus would cause it to form a chromatin loop and position the GULO pseudogene region inside a transcriptionally repressive chromatin complex called a Polycomb Body—hence the region is polycomb-repressed (Li et al. 2013).
Of particular importance to inactivating any aberrant transcription or influence of intact regulatory elements within the broken GULO locus, is the fact that this repression must be heritable and maintained over numerous cell divisions. In accord with this requirement, the region must be epigenetically tagged with markers that read the pre-existing repressed chromatin state, such as the trimethylation of histone H3 at lysine 27, otherwise known as H3K27me3 marks (Pirrotta 2012; Yuan et al. 2012). Indeed, the human GULO pseduogene contains an extensive H3K27me3 histone profile across the entire 28,800 base region that would faithfully maintain and recruit the defunct damaged GULO region for polycomb repression over succeeding cell divisions. These H3K27me3 histone marks are not only required for the long-term multigenerational marking of repressed regions, but they also stabilize the binding of the DNA to the polycomb repressive complex (Li et al. 2013; Pirrotta 2012).
Based on current ENCODE data, the dynamic and fault tolerant machinery of the human genome has recognized and epigenetically silenced any intact regulatory features within the GULO locus in a manner that is faithfully reproduced and protects the expression of neighboring genes. If the GULO locus was not effectively silenced, it is possible that neighboring genes would be negatively affected based on current models of chromatin functionality and gene regulation (Kapranov and St Laurent 2012; Van Bortle and Corces 2012). To the right (downstream) of the GULO locus is the CLU gene encoded on the minus strand, and to the left (upstream) is the EPHX2 gene encoded on the plus strand. Both genes are functional and actively expressed in the human genome (data not shown). So the GULO locus is the only gene targeted for epigenetic silencing in an otherwise transcriptionally active region of the chromosome.
Patterns of GULO Evolutionary Discontinuity In Humans and Apes
The GULO pseudogene has been promoted as supposed proof of human evolution from a common ancestor with apes. One recently popularized idea by Fairbanks claimed that, in regards to the broken GULO gene, “All primates have the same highly mutated fragment so the gene probably was disabled before different primate lineages diverged from their common ancestor” (Fairbanks 2010, p. 53). Fairbanks also claims that the human and chimpanzee version of the GULO pseudogene are 98% identical based on the blast results of a 565 base fragment from the gene, despite the fact that the entire GULO locus in humans, not counting the leading upstream sequence (~13,000 bases) associated with the apparent loss of several exons, is 28,800 bases long (fig. 2A). As we shall soon see, DNA similarity over the entire GULO region is markedly lower than Fairbanks biased estimate that used less than 2% of the total GULO pseudogene sequence.
Before discussing current research of the GULO gene region in humans, it is important to first note a recent research paper on this topic by two evolutionary scientists (Lachapelle and Drouin 2011). In this effort, the researchers attempted to ascertain the molecular basis for the apparent loss of five different exons in the human and ape GULO genes, compared to rodents. In particular, they analyzed the transposable element (TE) content of the various GULO gene regions since deletion-based inactivation of genes is largely associated with TEs and unequal recombination associated with their repeats (Teixeira-Silva et al. 2013; Uddin et al. 2006). Much to the researchers’ dismay, the overall TE content of mouse, rat, and guinea pig GULO genes varied widely and the TE content in humans and primates was not amenable to phylogenetic analyses due to its evolutionarily discordant nature. Despite the fact that TEs are apparently one of the main genomic drivers of deletion events in the genome, the researchers (Lachapelle and Drouin 2011) concluded that the lineage specific TE insertion patterns, which defied the standard inferred evolutionary model for primates, did not contribute to the loss of exons in the GULO gene. Thus, their evolutionary presuppositions caused the rejection of otherwise strong genomic data that implicated TE related unequal recombination at the GULO locus (resulting in exon deletion) that occurred in taxonomically restricted events.
The conclusions by Lachapelle and Drouin (2011) based on evolutionary presuppositions were similar to an earlier, but less exhaustive study by Nishikimi’s research group which showed that evolutionarily confusing patterns of TE fragments were present in exon-deletion areas of human and ape GULO pseudogenes (Inai, Ohta, and Nishikimi 2003; Nishikimi et al. 1994; Ohta and Nishikimi 1999). Clearly, the inactivated GULO gene and its TE content should be evaluated more closely in relation to the issue of human and primate origins, despite the lack of evolutionary common ancestry signatures.
Comparative Analysis of the Human and Ape GULO Genes
The reports of high DNA similarity across the GULO gene region in human, compared to apes are primarily based on the comparison of selected exon sequences and not the entire sequence (let alone regulatory regions) (Fairbanks 2010; Kawai et al. 1992; Nishikimi et al. 1994; Ohta and Nishikimi 1999). It is the high level of sequence conservation in these select exonic regions across diverse vertebrate taxa that strongly suggest that the human GULO pseudogene exons are the signature of a once viable and important gene (fig 2B). Compared to the 11 conserved exons of the consensus mammalian GULO gene, humans and apes have allegedly lost exons 1, 2, 5, 7, and 10 (Yang 2013). This does not include exon 1 of the rat GULO, which appears to be absent in other mammals.
The areas of putative exon deletion contain a wide variety of TEs and TE fragments (SINEs, LINEs, LTRs, and DNA elements). Many of these TE patterns are taxonomically restricted among humans, chimps, and gorillas in their presence and absence fragment signatures (fig 3A). I call these areas of interest “degenerate zones” or DZs (fig. 2A, fig. 3). There are four such areas corresponding to the putative loss of GULO exons. They are labeled DZ-1, DZ-2, DZ-3, and DZ-4 as shown in fig. 2. All degenerate zones except DZ-4, contain clear regions of genomic TE landscape differences that are taxonomically restricted, thus providing no discernible patterns supporting common ancestry of humans and apes. This is shown in fig. 3A and was noted previously in several evolutionary reports (Drouin, Godin, and Page 2011; Lachapelle and Drouin 2011).
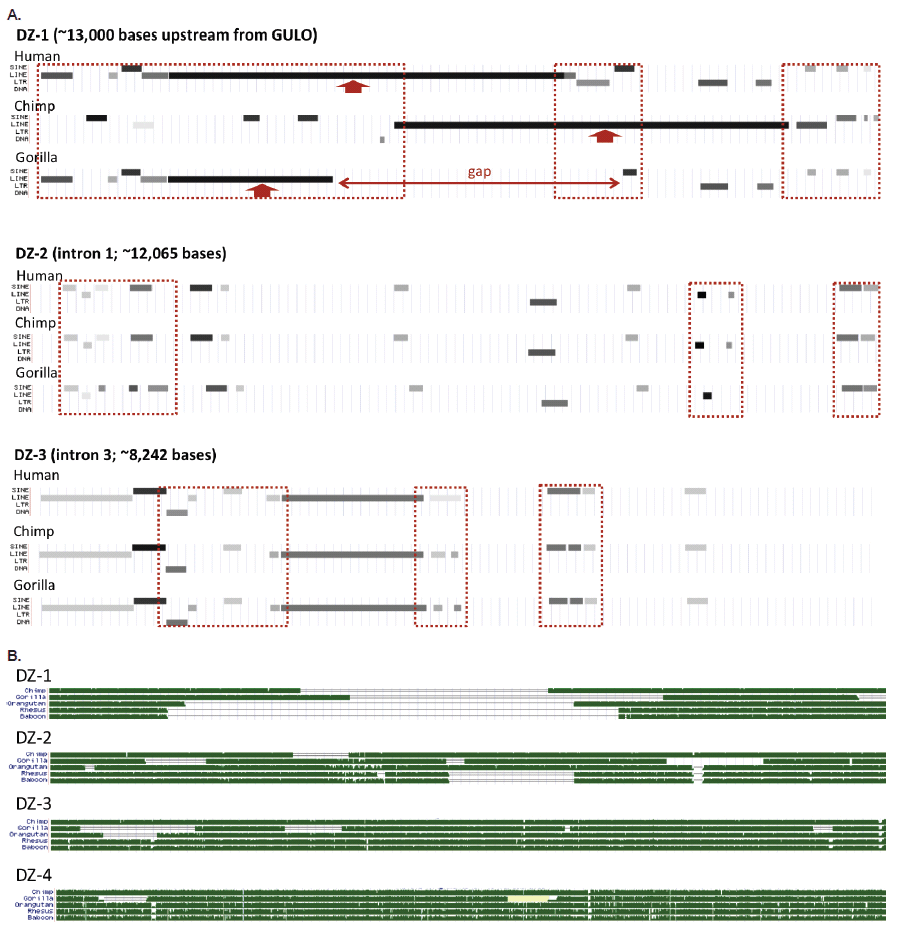
Fig. 3. Structure of the human GULO regions associated with putative exon loss demarcated as degenerate zones (DZs, see fig. 1). (A) Variable transposable element landscape of the first three degenerate zones for humans, chimps, and gorillas (DZ-4 showed no major differences). (B) Sequence homology tracks of five different ape taxon compared to each of the four human DZs in the GULO pseudogene. All data in this figure taken from the UCSC genome browser.
When the complete sequences of all four degenerate zones are compared between humans, chimps, gorillas, orangutans, rhesus macaque, and baboon, there are obvious taxonomically restricted gaps and deletion patterns that also provide a clear picture of discontinuity (fig. 3B).
What about the intact six GULO exons that are so highly conserved among vertebrates, several of which have been used as evidence of human-ape common ancestry? These human exon sequences actually vary widely in DNA sequence identity compared to five different ape taxa in the range of 74.8–100% (Table 2). However, when each group of human and ape exons is placed in similarity-ordered trees, the only set of sequences that produced the standard inferred phylogeny was for exon 5 (fig. 4A), which is the primary exon chosen by evolutionists as a target for comparative analyses (Nishikimi et al. 1994; Ohta and Nishikimi 1999; Truman and Borger 2007). The other exon sequences all produce markedly different trees that are decidedly taxonomically incongruent according to the inferred human-ape evolutionary tree. This pervasive evolutionarily discordant trend observed when constructing sequence-based trees, not only among humans and apes, but across the entire spectrum of eukaryotic life, is referred to as incomplete linage sorting (Tomkins and Bergman 2013).
Table 2. The DNA sequence identity for each human GULO exon compared to five different ape taxon (DNA sequences extracted from the UCSC genome browser). Identities include insertions and deletions.
| Percent Sequence Identity Compared to Human | ||||||
| Human GULO exon | Length (bases) | Chimp | Gorilla | Orangutan | Baboon | Rhesus |
| 1 | 101 | 99.1 | 100 | 97.1 | 91.1 | 91.1 |
| 2 | 96 | 98.0 | 98.0 | 94.8 | 92.8 | 88.6 |
| 3 | 107 | 98.2 | 85.0 | 98.2 | 74.8 | 74.8 |
| 4 | 151 | 99.4 | 90.8 | 97.4 | 94.1 | 92.8 |
| 5 | 164 | 97.6 | 97.0 | 94.6 | 91.5 | 90.3 |
| 6 | 129 | 97.7 | 98.5 | 99.3 | 95.4 | 93.8 |
Adding to the story of evolutionary discontinuity among the exon sequence comparisons, is the fact that single nucleotide polymorphisms (SNPs) in the human GULO exons are minimal (fig. 4B). Two exons have zero SNPs and the others have only 1 to 4 SNPs. The majority of human GULO SNPs are in the intronic regions. Thus, the evolutionarily discordant exon-based phylogenetic trees cannot be blamed on excessive sequence variability in these regions associated with the standard neutral model of evolution.
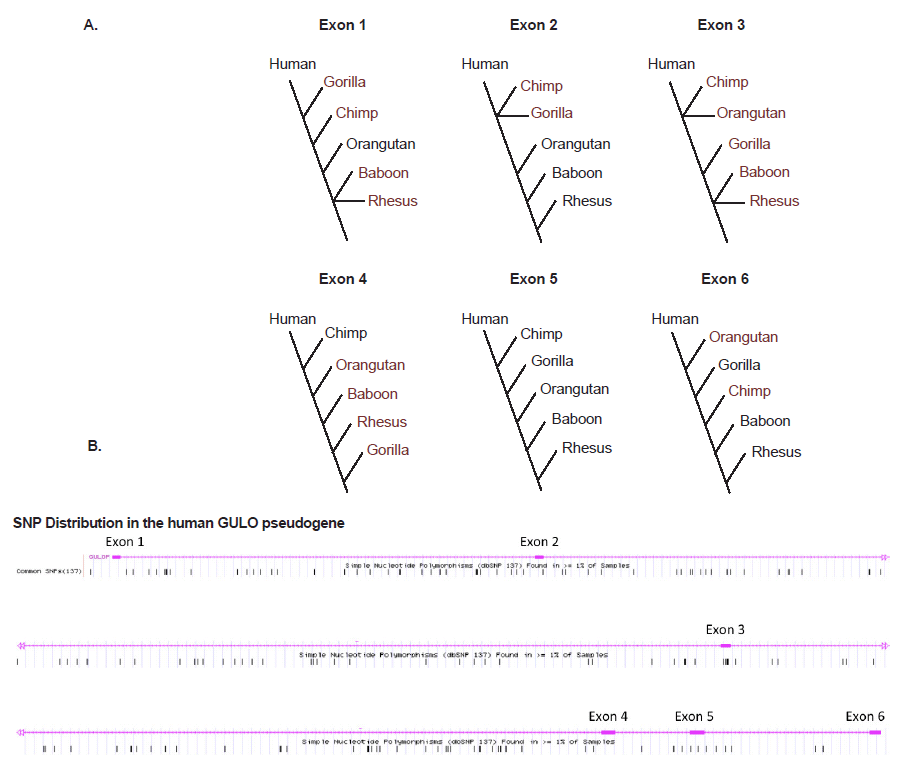
Fig. 4. (A) Simple DNA similarity trees for human and five ape taxon based on sequence identity of the six exons in the GULO pseudogene listed in Table 1. (B) Frequency of SNPs across the human GULO pseudogene (data from the UCSC genome browser).
Contrary to Fairbank’s claims of 98% human-chimp GULO similarity based on the comparison of a very small highly similar DNA fragment of 564 bases, the entire 28,800 base GULO region in human (hg19; chr8:27417791-27446590), which contains the putative remnants of six exons and five introns, is only 84% identical compared to chimpanzee using the previously established technique of optimized sequence slices and the BLASTN algorithm (Tomkins 2013b). Even more interesting is the comparison with gorilla using the same technique, which yielded 87% identity. These similarities are clearly outside the evolutionary paradigm and gorilla is more similar to human in the GULO region than chimpanzee—negating the inferred order of phylogeny.
A region comprising 13,000 bases upstream of the 6-exon human GULO sequence was also analyzed for similarity to chimpanzee and gorilla. Hypothetically, this upstream segment would correspond to the loss of the first two consensus mammalian exons in the GULO gene and perhaps contain remaining fragments of the gene promoter (fig. 2). Compared to chimp and gorilla, this segment was only 68% and 73% identical, respectively. Once again, these similarities are clearly outside the evolutionary paradigm and gorilla is much more similar in this upstream region than chimpanzee—negating the inferred order of phylogeny.
The comparative transposable element landscape of the upstream 13,000 base region demarcated as degenerate zone 1 (DZ-1), is characterized by multiple TE fragment differences between humans, chimps, and gorillas (fig. 3A). In a broader comparison which includes rhesus and baboon, the degradation of DZ-1 is clearly taxonomically restricted and evolutionarily discontinuous (fig. 3B).
Summary and Conclusions
One of the key arguments used by some proponents of evolutionary dogma is that the human and chimpanzee GULO pseudogenes are examples of inherited shared genetic mistakes. However, as demonstrated in this report, overall evidence for common ancestry across the entire human GULO locus with chimps and other apes is completely negated by the following discoveries:
- Compared to chimpanzee and gorilla, the 28,800 base GULO region in humans is only 84% and 87% identical to chimpanzee and gorilla, respectively.
- The 13,000 bases upstream of the human GULO region corresponding to the putative area of loss for at least two major exons, is only 68% and 73% identical to chimpanzee and gorilla, respectively.
- The individual six exon regions which are generally very similar among humans and various apes, each independently exhibit completely different and discordant phylogenetic patterns of similarity for all but one exon. A general lack of human SNPs in these exons indicates that incomplete lineage sorting based on alleged ancestral polymorphisms is not a likely explanation.
- The transposable element (TE) content for each region of putative exon loss is taxonomically restricted and evolutionarily discordant between humans and each ape taxon. Unequal recombination associated with transposable element repeats can lead to disruptive deletion events in the genome and likely did in these regions.
Many pseudogenes in the human genome are proving to be highly functional and key regulators of gene and genome function—producing important noncoding RNAs (Tomkins 2013a; Tomkins 2013c; Wen et al. 2012). However, there are other instances where pseudogenes likely represent broken genes that are tolerated by the organism, which would be expected under a model of genetic entropy (Sanford 2010). In the case of vitamin pathway genes across the spectrum of life, there are widespread documented instances where these seemingly unitary sequences have been broken and the lost vitamin is supplemented externally through diet or a host relationship (Helliwell, Wheeler, and Smith 2013).
However, the ability of an external source to supplement the biochemical loss of a vitamin should primarily be viewed as basic survival and a non-optimal situation involving toleration of the mutation event. In reality, the loss of a seemingly isolated gene sequence can have larger biochemical network effects and lower system optimization. In the study of mice who have had their entire GULO gene deleted from a naturally occurring mutation and are prone to spontaneous bone fractures (sfx mutants), at least 269 protein coding genes were found to be aberrantly regulated in diverse tissues even with vitamin C supplementation (Jiao et al. 2011). This is despite the fact that the mice appeared to grow normally and had no differences for internal organ weights, except for the spleen that weighed less than normal. Interestingly, a previous study had also detected the inability of vitamin C supplementation to negate certain physiological pathologies in sfx mutant mice (Lee et al. 2008). Clearly, adequate vitamin C intake can negate many problems associated with GULO loss, but the consequences may be less than optimal. Perhaps this mutation is in part, playing some role in the loss of human longevity as documented in the early chapters of Genesis in the Bible. Hypothetically, it could have occurred prior to the Genesis flood and then passed on as a pervasive trait in the genetic bottleneck in humans and various animals.
Another important aspect of consideration for the GULO gene degradation found in humans is the fact that it occurs frequently in mammals and birds, and the events typically are associated with complete or partial loss of exons. This indicates that the gene may be predisposed to the effects of genetic entropy. At present, exon degradation and deletion events in the GULO gene have been documented in humans, apes, pigs, rats, mice, guinea pigs, bats, and passerine birds (Cui et al. 2011a; Cui et al. 2011b; Drouin, Godin, and Page 2011; Harris et al. 2005; Hasan et al. 2004; Jiao et al. 2005; Kawai et al. 1992; Lachapelle and Drouin 2011; Nishikimi, Kawai, and Yagi 1992; Yang 2013). Thus, contrary to popular opinion promoted by a variety of evolutionary propagandists, GULO gene dysfunction appears to be a wide-spread feature among not just humans, apes, and guinea pigs, but terrestrial animal life in general. But most importantly, patterns of GULO DNA sequence degradation are taxonomically restricted and do not support the macro-evolutionary model of origins.
While the exon deletion events in both humans and apes appear to involve the same exons, the transposable element signatures surrounding the events are clearly non-similar and taxonomically restricted as noticed in this report and several others (Drouin, Godin, and Page 2011; Lachapelle and Drouin 2011). The question then arises as why the same exons were apparently deleted, even though the deletion events are unique, not only between humans and apes, but also between each of the different ape taxon. First, it should be noted that we really don’t know what the original genes looked like in their pristine created state or how similar they were between taxa. Second, there are several gaps in the ape GULO gene sequences and all ape genomes are still being assembled based on using the human genome as a framework (Prado-Martinez et al. 2013; Tomkins 2011). Thus, the ape genomes are biased to be more similar to human due to the way they are assembled and annotated.
Perhaps the anomaly that the same alleged exons were deleted is due to the fact that the transposable element features located in the intervening introns predisposed them to the effects of entropy. The GULO regions between human, chimpanzee, and gorilla do share similar genetic code—a common feature in engineered systems with similar function and it would be expected that the gene regions encoding common biochemical pathway enzymes would have a high level of similarity. Nevertheless, the apparent deletion signatures and remaining genetic landscapes are clearly not supportive of common ancestry. The same sort of GULO gene degradation has also been observed in many different passerine bird and bat taxon as well, and according to the opinion of this author, all represent different created kinds as do humans (created in the image of God) and the various types of ape taxons.
In 2007, Criswell proposed from a creationist perspective that the GULO pseudogene may serve some other functional purpose and not represent a degraded gene (Criswell 2007). However, he provided no analysis of the available human transcriptome data that was well developed and publicly available at the time to support this idea. He also stated that only four exons were in the human GULO gene, yet we now know that there at least six exons, and as documented in this report, closely match functional GULO exons in various animal taxa (Table 1). In addition, Criswell’s hypothesis was proposed just prior to the first public release of data from the ENCODE project.
As demonstrated in this report, the GULO region is clearly chromatin repressed in a heritable fashion and not transcriptionally active. Based on the current state of genomic information presented in this report, the bulk of evidence indicates that the human GULO gene is pseudogenized via deletions occurring as a result of unequal recombination associated with transposable element repeats. These exon deletion events are taxonomically restricted in humans and in each of the different ape species tested. And in combination with an extreme lack of sequence similarity over the entire GULO locus, utterly negate the idea of macroevolutionary common ancestry.
References
Assis R., and D. Bachtrog. 2013. Neofunctionalization of young duplicate genes in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 110, no. 43:17,409–17,414.
Birney E. C., R. Jenness, and K. M. Ayaz. 1976. Inability of bats to synthesise L-ascorbic acid. Nature 260, no. 5552:626–628.
Birney E. C., R. Jenness, and I. D. Hume. 1980. Evolution of an enzyme system: Ascorbic acid biosynthesis in monotremes and marsupials. Evolution 34, no. 2:230–239.
Chatterjee, I. B. 1973. Evolution and the biosynthesis of ascorbic acid. Science 182, no. 4118:1271–1272.
Chaudhuri, C. R. and I. B. Chatterjee. 1969. L-ascorbic acid synthesis in birds: phylogenetic trend. Science 164, no. 3878:435–436.
Combs, G. F. Jr., 2008. The vitamins: Fundamental aspects in nutrition and health. San Diego, California: Elsevier.
Criswell, D. 2007. Adam and Eve, vitamin C, and pseudogenes. Acts & Facts 36, no. 5. Dallas, Texas: Institute for Creation Research.
Cui, J., Y. H. Pan, Y. Zhang, G. Jones, and S. Zhang. 2011a. Progressive pseudogenization: Vitamin C synthesis and its loss in bats. Molecular Biology and Evolution 28, no. 2:1025–1031.
Cui J, X. Yuan, L. Wang, G. Jones, and S. Zhang. 2011b. Recent loss of vitamin C biosynthesis ability in bats. PloS One 6, no. 11:e27114.
Drouin. G., J.-R. Godin, and B. Page. 2011. The genetics of vitamin C loss in vertebrates. Current Genomics 12, no. 5:371–378.
Fairbanks, D. J. 2010. Relics of Eden: The powerful evidence of evolution in human DNA. Amherst, New York: Prometheus Books.
Fu, W., T. D. O’Connor, G. Jun, H. M. Kang, G. Abecasis, S. M. Leal, S. Gabriel, et al. 2013. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, no. 7431:216–220.
Funk, C. 1912. The substance from yeast and certain foodstuffs which prevents polyneuritis (beri-beri). British Medical Journal II:787.
Gropper, S. S., and J. L. Smith. 2012. Advanced nutrition and human metabolism. Stamford, Connecticut: Cengage Learning.
Hancock, R. D., J. R. Galpin, and R. Viola. 2000. Biosynthesis of L-ascorbic acid (vitamin C) by Saccharomyces cerevisiae. FEMS Microbiology Letters 186, no. 2:245–250.
Harris, B. S., P. F. Ward-Bailey, L. R. Donahue, K. R. Johnson, R. T. Bronson, and M. T. Davisson. 2005. Spontaneous fracture 2 Jackson (sfx2J), a second mutation of the gulonolactone oxidase gene (Gulo). Retrieved from http://www.informatics.jax.org/allele/MGI:3510598.
Hasan, L., P. Vögeli, P. Stoll, S. S. Kramer, G. Stranzinger, and S. Neuenschwander. 2004. Intragenic deletion in the gene encoding L-gulonolactone oxidase causes vitamin C deficiency in pigs. Mammalian Genome 15, no. 4:323–333.
Helliwell, K. E., G. L. Wheeler, and A. G. Smith. 2013. Widespread decay of vitamin-related pathways: Coincidence or consequence? Trends in Genetics 29, no. 8:469–478.
Inai Y., Y. Ohta Y., and M. Nishikimi. 2003. The whole structure of the human nonfunctional L-gulono-gammalactone oxidase gene—the gene responsible for scurvy—and the evolution of repetitive sequences thereon. Journal of Nutritional Science and Vitaminology 49, no. 5:315–319.
Innan, H., and F. Kondrashov. 2010. The evolution of gene duplications: Classifying and distinguishing between models. Nature Reviews Genetics 11, no. 2:97–108.
Jiao, Y., X. Li, W. G. Beamer, J. Yan, Y. Tong, D. Goldowitz, B. Roe, and W. Gu. 2005. A deletion causing spontaneous fracture identified from a candidate region of mouse Chromosome 14. Mammalian Genome 16, no. 1:20–31.
Jiao, Y., J. Zhang, J. Yan, J. Stuart, G. Gibson, L. Lu, R. Willaims, and Y. J. Wang. 2011. Differential gene expression between wild-type and Gulo-deficient mice supplied with vitamin C. Genetics and Molecular Biology 34, no. 3:386–395.
Kapranov, P., and G. St Laurent. 2012. Dark matter RNA: Existence, function, and controversy. Frontiers in Genetics 3:60.
Kawai, T., M. Nishikimi, T. Ozawa, and K. Yagi. 1992. A missense mutation of L-gulono-gamma-lactone oxidase causes the inability of scurvy-prone osteogenic disorder rats to synthesize L-ascorbic acid. Journal of Biological Chemistry 267, no. 30:21,973–21,976.
Lachapelle, M. Y., and G. Drouin. 2011. Inactivation dates of the human and guinea pig vitamin C genes. Genetica 139, no. 2:199–207.
Lee, C. W., X. D. Wang, K. L. Chien, Z. Ge, B. H. Rickman, A. B. Rogers, A. Varro, M. T. Whary, T. C. Wang, and J. C. Fox. 2008. Vitamin C supplementation does not protect L-gulonogamma-lactone oxidase-deficient mice from Helicobacter pylori-induced gastritis and gastric premalignancy. International Journal of Cancer 122, no. 5:1068–1076.
Li, H. B., K. Ohno, H. Gui, and V. Pirrotta. 2013. Insulators target active genes to transcription factories and polycomb-gulonogammarepressed genes to polycomb bodies. PLoS Genetics 9, no. 4:e1003436.
Linster, C. L., T. A. Gomez, K. C. Christensen, L. N. Adler, B. D. Young, C. Brenner, and S. G. Clarke. 2007. Arabidopsis VTC2 encodes a GDP-L-galactose phosphorylase, the last unknown enzyme in the Smirnoff-Wheeler pathway to ascorbic acid in plants. Journal of Biological Chemistry 282, no. 26:18,879–18,885.
Linster, C. L., and E. Van Schaftingen. 2007. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS Journal 274, no. 1:1–22.
Martinez del Rio, C. 1997. Can passerines synthesize vitamin C? The Auk 114, no. 3:513–516.
Mohan, S., A. Kapoor, A. Singgih, Z. Zhang, T. Taylor, H. Yu, R. B. Chadwick, et al. 2005. Spontaneous fractures in the mouse mutant sfx are caused by deletion of the gulonolactone oxidase gene, causing vitamin C deficiency. Journal of Bone and Mineral Research 20, no. 9:1597–1610.
Nishikimi, M., R. Fukuyama, S. Minoshima, N. Shimizu, and K. Yagi. 1994. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. Journal of Biological Chemistry 269, no. 18:13,685–13,688.
Nishikimi, M., T. Kawai, and K. Yagi. 1992. Guinea pigs possess a highly mutated gene for L-gulono-gamma-lactone oxidase, the key enzyme for L-ascorbic acid biosynthesis missing in this species. Journal of Biological Chemistry 267, no. 30:21,967–21,972.
Nowak, G., C. A. Carter, and R. G. Schnellmann. 2000. Ascorbic acid promotes recovery of cellular functions following toxicant-induced injury. Toxicology and Applied Pharmacology 167, no. 1:37–45.
Ohta, Y., and M. Nishikimi. 1999. Random nucleotide substitutions in primate nonfunctional gene for L-gulonogamma-lactone oxidase, the missing enzyme in L-ascorbic acid biosynthesis. Biochimica et Biophysica Acta 1472, no. 1–2:408–411.
Padh, H. 1990. Cellular functions of ascorbic acid. Biochemistry and Cell Biology 68, no. 10:1166–1173.
Phillips, J. E., and V. G. Corces. 2009. CTCF: Master weaver of the genome. Cell 137, no. 7:1194–1211.
Pirrotta, V. 2012. Molecular biology. How to read the chromatin past. Science 337, no. 6097:919–920.
Pohanka, M., J. Pejchal, S. Snopkova, K. Havlickova, J. Z. Karasova, P. Bostik, and J. Pikula. 2012. Ascorbic acid: An old player with a broad impact on body physiology including oxidative stress suppression and immunomodulation: A review. Mini Reviews in Medicinal Chemistry 12, no. 1: 35–43.
Prado-Martinez, J., P. H. Sudmant, J. M. Kidd, H. Li, J. L. Kelley, B. Lorente-Galdos, K. R. Veeramah, et al. 2013. Great ape genetic diversity and population history. Nature 499, no. 7459:471–475.
Roy, R. N., and B. C. Guha. 1958. Species difference in regard to the biosynthesis of ascorbic acid. Nature 182, no. 4631: 319–320.
Sanford, J. C. 2010. Genetic entropy and the mystery of the genome. Waterloo, New York: FMS Publications. Smirnoff, N. 2001. L-ascorbic acid biosynthesis. Vitamins and Hormones 61:241–266.
Teixeira-Silva, A., R. M. Silva, J. Carneiro, A. Amorim, and L. Azevedo. 2013. The role of recombination in the origin and evolution of alu subfamilies. PloS One 8, no. 6:e64884.
Tennessen, J. A., A. W. Bigham, T. D. O’Connor, W. Fu, E. E. Kenny, S. Gravel, S. McGee, et al. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, no. 6090:64–69.
Tomkins, J. P. 2011. How genomes are sequenced and why it matters: Implications for studies in comparative genomics of humans and chimpanzees. Answers Research Journal 4:81–88. Retrieved from http://www.answersingenesis.org/articles/arj/v4/n1/implications-for-comparative-genomics.
Tomkins, J. P. 2013a. Alleged human chromosome 2 “fusion site” encodes an active DNA binding domain inside a complex and highly expressed gene—negating fusion. Answers Research Journal 6:367–375. Retrieved from http://www.answersingenesis.org/articles/arj/v6/n1/human-chromosome-fusion.
Tomkins, J. P. 2013b. Comprehensive analysis of chimpanzee and human chromosomes reveals average DNA similarity of 70%. Answers Research Journal 6:63–69. Retrieved from http://www.answersingenesis.org/articles/arj/v6/n1/human-chimp-chromosome.
Tomkins, J. P. 2013c. The human beta-globin pseudogene is non-variable and functional. Answers Research Journal 6:293–301. Retrieved from http://www.answersingenesis.org/articles/arj/v6/n1/human-beta-globin-pseudogenes.
Tomkins, J., and J. Bergman. 2013. Incomplete lineage sorting and other ‘rogue’ data fell the tree of life. Journal of Creation 27, no. 3:84–92.
Truman, R., and P. Borger. 2007. Why the shared mutations in the Hominidae exon X GULO pseudogene are not evidence for common descent. Journal of Creation 21, no. 3:118–127.
Uddin, R. K., Y. Zhang, V. M. Siu, Y.-S. Fan, R. L. O’Reilly, J. Rao, and S. M. Singh. 2006. Breakpoint associated with a novel 2.3 Mb deletion in the VCFS region of 22q11 and the role of Alu (SINE) in recurring microdeletions. BMC Medical Genetics 7:18.
Van Bortle, K., and V. G. Corces. 2012. Nuclear organization and genome function. Annual Review of Cell and Developmental Biology 28:163–187.
Wen, Y. Z., L. L. Zheng, L. H. Qu, F. J. Ayala, and Z. R. Lun. 2012. Pseudogenes are not pseudo any more. RNA Biology 9, no. 1:27–32.
Wong, S. Z. H., B. Ching, Y. R. Chng, W. P. Wong, S. F. Chew, and Y. K. Ip. 2013. Ascorbic acid biosynthesis and brackish water acclimation in the Euryhaline Freshwater White-Rimmed Stingray, Himantura signifer. PloS One 8, no. 6:e66691.
Yang, H. 2013. Conserved or lost: Molecular evolution of the key gene GULO in vertebrate vitamin C biosynthesis. Biochemical Genetics 51, no. 5–6:413–425.
Yang, L., M. Zou, B. Fu, and S. He., 2013. Genome-wide identification, characterization, and expression analysis of lineage-specific genes within zebrafish. BMC Genomics 14:65.
Yuan, W., T. Wu, H. Fu, C. Dai, H. Wu, N. Liu, X. Li, et al. 2012. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 337, no. 6097:971–975.