The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
A major argument supposedly supporting human evolution from a common ancestor with chimpanzees is the “chromosome 2 fusion model” in which ape chromosomes 2A and 2B purportedly fused end-to-end, forming human chromosome 2. This idea is postulated despite the fact that all known fusions in extant mammals involve satellite DNA and breaks at or near centromeres. In addition, researchers have noted that the hypothetical telomeric end-to-end signature of the fusion is very small (~800 bases) and highly degenerate (ambiguous) given the supposed 3 to 6 million years of divergence from a common ancestor. In this report, it is also shown that the purported fusion site (read in the minus strand orientation) is a functional DNA binding domain inside the first intron of the DDX11L2 regulatory RNA helicase gene, which encodes several transcript variants expressed in at least 255 different cell and/or tissue types. Specifically, the purported fusion site encodes the second active transcription factor binding domain in the DDX11L2 gene that coincides with transcriptionally active histone marks and open active chromatin. Annotated DDX11L2 gene transcripts suggest complex post-transcriptional regulation through a variety of microRNA binding sites. Chromosome fusions would not be expected to form complex multi-exon, alternatively spliced functional genes. This clear genetic evidence, combined with the fact that a previously documented 614 Kb genomic region surrounding the purported fusion site lacks synteny (gene correspondence) with chimpanzee on chromosomes 2A and 2B (supposed fusion sites of origin), thoroughly refutes the claim that human chromosome 2 is the result of an ancestral telomeric end-to-end fusion.
Keywords: Chromosome 2 fusion, DDX11L2 gene, human evolution, human-chimpanzee
Introduction
One of the most common arguments for human evolution is the hypothetical head-to-head fusion of two small acrocentric ape-like chromosomes to form human chromosome 2. This is thought to account for the discrepancy in chromosome numbers between humans and apes, who have 46 and 48 chromosomes, respectively (Yunis and Prakash 1982). This original hypothesis was based on shared banding patterns observed under light microscopy for stained mitotic chromosomes. In regard to the inferred fusion, the ends located on the short arms of what are now called chimpanzee chromosomes 2A and 2B were thought to have fused in an end-to-end model. For a depiction of the fusion event using comparatively scaled drawings of cytogenetic images, see Fig. 1. For a recent review of the literature see Bergman and Tomkins (2011) and for a recent re-analysis of the fusion site and the putative cryptic centromere site, see Tomkins and Bergman (2011a).
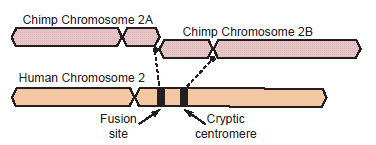
Fig. 1. Depiction of a hypothetical scenario in which chimpanzee chromosomes 2A and 2B fused end-to-end to form human chromosome 2. All chromosomes were comparatively drawn to scale according to cytogenetic images by Yunis and Prakash (1982). Putative fusion and cryptic centromere sites were placed on human chromosome 2 based on current locations in the UCSC Genome Browser. Note the extreme lack of positional correspondence for the cryptic centromere site and to a lesser extent, the fusion site and the current human centromere. Also note the size discrepancy which is about 10% or 24 million bases.
If indeed the DNA sequence pattern in this region indicated a true chromosomal end-to-end fusion event, then it would be the first such case identified in mammals that involved a telomere-to-telomere fusion. This is due to the fact that in all documented extant mammalian chromosome fusions to date, satellite DNA (satDNA) is a key genomic feature comprising the breakage and subsequent fused sequence (Adega, Guedes-Pinto, and Chaves 2009; Chaves et al. 2003; Tsipouri et al. 2008). In such cases, the junction is either demarcated by telomere-satDNA or satDNA-satDNA, typically involving centromeres or regions proximal to them. Chromosome fusions representing telomere-telomere signatures are not presently documented, except for the hypothetical fusion of human chromosome 2. This absence of documented end-to-end telomere fusions in living mammals is largely due to the fact that telomeres contain a highly specialized end cap called the shelterin protein complex that protects them from fusion (Tomkins and Bergman 2011b).
It is noteworthy that centromeric satellite DNA was originally suspected as playing a role in the break point and fusion mechanism for the hypothetical human chromosome fusion 2 event since a significant amount of the chimpanzee DNA on chromosome 2B cannot be accounted for in the telomere-to-telomere fusion model (Ijdo et al. 1991). Based on the current genome assembly for human chromosome 2 at ~243 Mb (hg19; genome.ucsc.edu), and the determination of about 10% chromosome loss in chimpanzee chromosome 2B based on comparative cytogenetics (Yunis and Prakash 1982), approximately 24 Mb of DNA would have been lost in the fusion (fig. 1).
In a recent analysis characterizing the DNA sequence and genomic organization of the chromosomal telomere end cap regions of gorilla and chimpanzee chromosomes, researchers found large amounts of chimpanzee specific satDNA at the ends of chromosomes 2A and 2B, but absent in human (Ventura et al. 2012). The authors postulated that the chimp satDNA was deleted in the fusion event, yet the telomere sequence distal to it, somehow remained. Despite the proposed deletions of DNA in the fusion event described by Ventura et al., their model cannot realistically account for a 24 Mb DNA loss.
The idea of a head-to-head telomeric fusion first emerged when a putative fusion site was cloned and sequenced, showing a signature of about 800 bases in length on human chromosome two in region 2q13 (Ijdo et al. 1991). In 2002, researchers completely sequenced and annotated ~614 Kb of DNA surrounding the fusion site (Fan et al. 2002a; Fan et al. 2002b). The two studies published by Fan et al. brought to light a number of serious problems that completely contradicted the fusion model, discussed in turn below.
First, was the problem of lack of synteny (corresponding gene content and DNA sequence similarity) with chimpanzee surrounding the putative fusion region on human chromosome 2. In addition to the unaccounted for extreme loss of chimp DNA in the hypothetical fusion, the putative fusion site was surrounded by a wide array of functional genes and putative pseudogenes with no homology to the ends of chimpanzee chromosomes 2A or 2B, their supposed ancestral sites of origin. Since the researchers could not find any similarity with chimpanzee for the gene content surrounding the putative fusion site, they postulated that the genes were transferred from other parts of the human genome after the fusion event occurred.
Second, the putative fusion sequence is highly degenerate given the inferred evolutionary timescale. In their paper, Fan et al. (2002a, p. 1657) state “Only 48% of the 127 repeats in RP11–395L14 and 46% of the 158 repeats in M73018 are perfect TTAGGG or TTGGGG units” and “If the fusion occurred within the telomeric repeat arrays less than ~6 Mya, why are the arrays at the fusion site so degenerate?” Tomkins and Bergman reevaluated the degeneracy of the fusion site along with the possible presence of other telomere repeats in a 177 Kb region surrounding it and found that not only was the putative fusion sequence itself ambiguous, but restricted to a single region of only about 800 bases in length (Tomkins and Bergman 2011a).
Third, one of the most remarkable discoveries about the putative fusion site by Fan et al. (2002b) was its location inside a CHLR1-like pseudogene (now called DDX11L2) as shown in Fig. 1 of their report (Fan et al. 2002b, p. 1664). However, the text of their report did not specifically discuss its anomalous location inside the pseudogene, despite the fact that their graphical annotation clearly showed that it was. Since 2002, this region of the human genome has been updated with improved annotations as well as a significant amount of unpublished publicly available ENCODE data.
As demonstrated in this report, the purported fusion site encodes an active transcription factor binding site and is definitively located inside the first intron of a functional RNA helicase gene transcribed on the minus strand. The location of the putative fusion sequence inside a functional and highly expressed gene associated with a wide variety of cellular processes strongly negates the idea that it is the by-product of a hypothetical head-to-head telomeric fusion.
The Fusion Motif Encodes a Functional Domain in the DDX11L2 Gene
As initially reported by Fan et al. (2002b), the putative 800 base fusion sequence is located somewhere inside a CHLR1 pseudogene within human chromosome region 2q13–2q14.1. The CHLR1 type category of genes in humans was originally annotated and characterized based on the DEAD family of DNA and RNA helicase genes, first discovered in yeast and found to be critical for proper chromosome transmission during mitosis (Gerring, Spencer, and Hieter 1990), and then eventually studied in humans (Abdelhaleem, Maltais, and Wain 2003; Cordin et al. 2006). The DEAD genetic acronym stands for the abbreviations of the amino acids associated with the key functional motif, the DEAD-box [asparagine (D), glutamic acid (E), alanine (A), asparagine (D)]. The DEAD-box helicases are thought to be enzymes that catalyze the separation and manipulation of nucleic acid polymers in an energy-dependent manner (Abdelhaleem, Maltais, and Wain 2003; Cordin et al. 2006).
Since the original complete sequencing of the fusion region on chromosome 2 (Fan et al. 2002a), the gene containing the fusion sequence has since been renamed from CHLR1 to DDX11L2 and found to be a member of the DDX11L family of at least 18 different RNA helicase genes (Costa et al. 2009). Oddly, while Costa et al. functionally and structurally characterized the DDX11L2 gene, they mentioned nothing of the fact that it contained the well-known chromosome 2 fusion sequence. Because the evolutionary model of gene origins is largely based on the idea of duplication from an original ancestral sequence, Costa et al. proposed that the variants of DDX11L genes in humans all evolved from ancestral sequences in apes. However, when a human DDX11L gene sequence was used as a cytogenetic probe for fluorescence in situ hybridization (FISH) in chimpanzee, it only hybridized to two places on chimp chromosomes 12 and 20 [image url: http://www.biomedcentral.com/1471-2164/10/250/figure/F3]. The same FISH experiment was also done in gorilla and showed four areas of gene synteny on chromosomes 3, 6, 7, and 20. In complete contradiction to evolutionary predictions, the human DDXL11L gene showed no synteny with chromosomes 2A or 2B in chimpanzee or gorilla (see image url above). This is highly significant because as described below, the fusion site appears to be a key functional motif contained within the DDX11L2 gene on chromosome 2. Furthermore, the fact that 18 copies of the DDX11L gene exists in humans verses only two copies in chimps and four in gorillas, is completely discordant with the inferred human-ape evolutionary phylogeny. Another evolutionary discordant fact about these genes is that their genomic locations are all different in each of the human and ape genomes.
Based on the most recent annotation of the human genome (GRCh37/hg19; http://genome.ucsc.edu), the ~800 base purported fusion site is clearly contained within the first intron of the DDXL11L2 gene on human chromosome two as depicted in Fig. 2A, 2B. The DDXL11L2 gene is composed of three primary exons and is transcribed from the telomere to centromere direction on the minus strand (fig. 2A, 2B). Thus, the so-called fusion sequence is actually read (5' to 3') in the reverse complement as part of a functional gene, not the forward strand orientation as typically depicted by the so-called fusion signature sequence (Fairbanks 2010; Tomkins and Bergman 2011a). Additionally, the fusion site contains data tracks for transcription factor binding (fig. 2A, 2B) indicating that it contains a functional DNA binding domain. Specifically, the three transcription factors CTCF, cMyc, and Po12 have been shown to bind to the putative fusion region in chromatin immunoprecipitation DNA sequencing (ChIP-seq) studies.
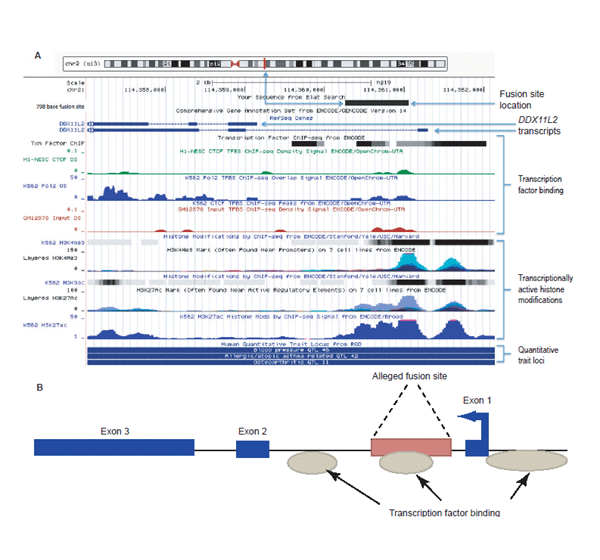
Fig. 2. (A) UCSC genome browser data showing selected gene annotation and ENCODE-related tracks for the DDX11L2 gene locus with the 798 base fusion site positioned within the locus using BLAT. Analysis image accessed at genome.ucsc.edu on July 23, 2013. (B) Simplified graphic showing the fusion site inside the DDX11L2 gene for the full-length transcript. Arrow in first exon depicts direction of transcription. (Click image for larger view)
There are actually three regions of consensus transcription factor binding in the DDXL11L2 gene with the two strongest regions of binding occurring in the fusion site and also directly 5' and proximal to the first exon in the gene’s promoter region. These two main areas of transcription factor binding coincide with specific epigenetic markers associated with transcriptional activity (fig. 2A, 2B). Of particular importance is the extensive combinatorial presence of specific transcriptionally active histone marks associated with acetylation (H3K27ac, H3K9ac) and methylation-based (H3K4Me1, H3K4Me3) modifications identified across the fusion site and the genes promoter area. These transcriptionally active epigenetic chromatin marks coincide with the areas of transcription factor binding. Combined with the evidence for transcription factor binding domains, the combinatorial histone marks clearly demarcate these regions as transcriptionally active and key to the expression of the DDXL11L2 gene. Interestingly, the H3K27ac histone acetylation marks are also typically associated with active enhancer elements in long-range chromatin interactions associated with transcription (Creyghton et al. 2010; Zentner, Tesar, and Scacheri 2011) and, of course, associated with active gene promoters (Dunham et al. 2012; Harmston and Lenhard 2013).
DDXL11L2 Transcripts are Variable and Complex
If the purported fusion site sequence actually represents a DNA binding site motif and is key to the function and expression of the DDXL11L2 gene, what types of transcripts are produced and how would a second promoter site in the first intron of the gene be important in the process? The UCSC Genome Browser shows two consensus transcripts, for the DDXL11L2 gene, both of which contain three exons, but only one contains the first exon directly 5' to (in front of) the intron containing the putative fusion sequence. In the the NCBI nucleotide database, these DDX11L2 gene sequences correspond to accessions NR_024005.2 and NR_024004.1), with RNA transcript lengths of 1,668 and 2,158 nucleotides, respectively. The longest transcript of 2,158 bases maps to the entire length of the DDXL11L2 gene (fig. 2A, 2B).
The DDX11L2 mRNA transcripts were annotated using the RegRNA2.0 computational platform (Chang et al. 2013) to look for functional RNA motifs that would give clues about their possible post-transcriptional regulation. Both DDX11L2 transcripts contained the same set of six different micro-RNA (miRNA) binding sites, with the longer transcript containing two additional sites. Micro-RNAs are small regulatory RNAs about 22 nucleotides in length that post-transcriptionally regulate both protein and non-protein coding genes via complementary binding to miRNA binding sites in gene transcripts. It is now believed that miRNAs are involved in virtually every cell process and regulate well over half of the human transcriptome (Pasquinelli 2012).
The diversity and combinatorial nature of miRNA binding sites (also called miRNA response elements) in RNA transcripts is hypothesized to represent the “Rosetta Stone” of a molecular language of regulatory communication in the transcriptome (Salmena et al. 2011). Thus, shared miRNA response elements among transcripts within gene families in conjunction with coexpression data, is a strong indicator of coordinated and co-regulated gene expression between a pseudogene and its protein coding homolog. Pseudogenes are now being proven to regulate their protein coding counterparts in complex coordinated networks of competitive complementary binding in conjunction with not only the protein coding transcripts themselves, but other miRNA interacting factors such as circular RNAs (Taulli, Loretelli, and Pandolfi 2013). This idea is highly significant for the current study, because when the five different transcript variants for the DDX11 protein coding gene were also analyzed for miRNA binding sites that might be shared with the DDX11L2 pseudogene, they contained two sites in common (hsa-miR-661 and hsa-miR-4739). As discussed below, the two genes also share common categorical tissue expression profiles and are both significantly coexpressed across multiple data sets.
DDXL11L2 is Highly Expressed and Coexpressed With DDX11
The BioGPS.org (Wu, Macleod, and Su 2013) gene annotation and analysis portal listed significant levels of expression data for the DDXL11L2 gene in 16 out of the 31 major tissue types found in the human body (fig. 3). Because, pseudogenes have been shown to produce transcripts involved in the regulation of their protein coding homologs, expression patterns for the DDX11 protein coding gene were also queried at BioGPS.org. Both DDXL11L2 and its homolog, DDX11, were found to be expressed in the same major tissue categories (fig 3). In addition, the genevestigator.com biomedical database listed significant levels of gene expression for DDX11L2 in 255 different cell and/or tissues types (Hruz et al. 2008).
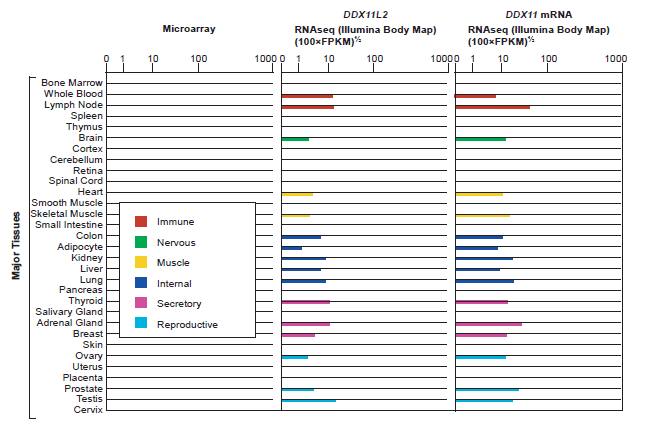
Fig. 3. BioGPS listing of gene expression by major tissue category for the DDX11L2 pseudogene and the DDX11 protein coding gene. Expression tissue categories are identical for both genes. In the CoExpress database (v5; http://coxpresdb.jp), both genes are significantly co-expressed together across multiple experimental data sets.
In the COXPRESSdb database (Obayashi et al. 2013), the DDXL11L2 gene is coexpressed at high levels with a variety of different blood cell development and chromatin remodeling genes, including linked expression with the protein coding DDX11 gene (ranked at 27 in the coexpression index with DDX11L2). As mentioned above, this coexpression data provides possible clues concerning the regulatory role of DDXL11L2 transcripts in association with DDX11 protein coding transcripts.
Clues about what type of general cellular processes the DDXL11L2 pseudogene might be involved with were also revealed through its detected close association with other genes. The highest levels of coexpression for DDX11L2 were directly linked to three different key genes:
- TREML1—a cell surface receptor in myeloid cells (non-lymphocyte blood cells—platelets) family,
- TUBB1—a member of the beta tubulin protein family specifically expressed in platelets and megakaryocytes (bone marrow cells responsible for production of blood platelets), and
- BEND2—a gene that encodes a protein with two domains associated with protein and DNA interactions that occur during chromatin restructuring and transcription.
The association with BEND2 is particularly interesting because the DDX11 helicase class of genes are associated with chromatin remodeling (Abdelhaleem et al. 2003; Cordin et al. 2006). Of the top 100 genes significantly coexpressed with DDX11L2, 27 had assigned KEGG (Kyoto Encyclopedia of Genes and Genomes) functions involving cell surface receptor interactions with the extracellular matrix, including the development of blood cells (table 1). Many of the other genes that were significantly coexpressed with DDX11L2 have not been assigned functional profiles yet by the research community.
Table 1. High–level functions and pathways for genes coexpressed with DDX11L2, the pseudogene containing the putative chromosome 2 fusion site. Data below represents information from the top 100 coexpressed genes for which a KEGG (Kyoto Encyclopedia of Genes and Genomes) description exists.
| KEGG ID | Title | #genes |
| hsa04062 | Chemokine signaling pathway (chemokines act as chemo-attractants guiding cell migration) | 6 |
| hsa04512 | ECM-receptor interaction (cell surface receptors that interact with the extra cellular matrix) | 6 |
| hsa04060 | Cytokine-cytokine receptor interaction (cell surface receptor interactions associated with small signaling molecules called cytokines) | 6 |
| hsa04640 | Hematopoietic cell lineage (blood cell development) | 5 |
| hsa04144 | Endocytosis (energy dependent process whereby cells absorb molecules–mediated by cell surface receptor interactions) | 4 |
Discussion
A putative, but degenerate head-to-head telomere fusion-like sequence of about 800 bases is one of the key pieces of evidence used by evolutionists to support the human chromosome 2 fusion model of two smaller ape-like chromosomes. However, the DNA sequence features do not match evolutionary expectations, being surprisingly small in size and extremely degenerate (Fan et al. 2002a; Tomkins and Bergman 2011a). In addition, the putative fusion site is not characterized by the presence of satellite DNA, a hallmark of known fusion events in living mammals, which was a surprise to researchers who first discovered it (Ijdo et al. 1991).
Interestingly, chimpanzee chromosome end caps are rich in a type of satellite DNA specific to chimpanzee sub-telomeric regions, including the ends of chromosomes 2A and 2B (Ventura et al. 2012).Yet none of this chimp end cap satDNA is located in the human genome, much less on chromosome 2 near the fusion site. Evolutionists have attempted to explain this anomaly by suggesting that the chimp-specific satDNA has been somehow eliminated over the course of human evolution and expanded in chimpanzee, a purely ad hoc explanation (Ventura et al. 2012). In fact, the chromosomal end cap DNA composition of humans, chimpanzees, gorillas, and orangutans has recently been found to be species-specific—representing a type of taxonomically restricted DNA sequence (Ventura et al. 2012). In a creationist model of origins, taxonomically restricted DNA sequences, are clear DNA-based evidence for all of these different types of apes and humans, being created uniquely after their kind. Clearly, the end cap DNA regions of humans and apes shows no evidence of descending along a lineage of common ancestry, much less fusing in a human-chimp common ancestor to form a new chromosome.
Remarkably, Ventura et al. (2012) go through an incredibly convoluted and complex hypothetical model to try and explain their primate evolution negating results in light of the sacred idea of a chromosome fusion. To explain the lack of chimp satDNA in human near the alleged fusion site, they claim that the satDNA was selectively deleted during the fusion event, while portions of the telomeric sequence were preserved. They also claim that a large section of DNA at the end of chimp chromosome 2B, which strangely had some homology to an internal region of human chromosome 10, was also deleted out in the process.
Not only does the end cap composition of ape and human chromosomes nullify the concept of a fusion event to produce human chromosome 2, but so does the extreme lack of genetic synteny surrounding the purported fusion site on human chromosome 2. As mentioned previously, a 614 Kb region surrounding the fusion site was sequenced and annotated showing a large number of genes and pseudogenes surrounding the alleged fusion sequence, all of which had no synteny to chimpanzee chromosomes 2A or 2B, their supposed ancestral sites of origin (Fan et al. 2002b). Instead of capitulating on the idea of an evolutionary fusion, the authors postulated that the gene neighborhood surrounding the purported fusion site was derived by duplication and copying from other genes and regions around the human genome. Amazingly, the authors also noted the presence of the fusion site as being located inside a putative RNA helicase pseudogene in one of their figures, but minimized the evidence of the discovery in the text of their report (Fan et al. 2002b).
Ultimately, the fact that the fusion site is located inside a possibly important and functional gene is inconsistent with the hypothesis that it arose from some sort of major genetic aberration, such as a chromosomal fusion. In this report, the purported fusion site and its remarkable presence inside a clearly active gene is revisited with fresh data mined from the human genome project combined with gene expression data available in a variety of public databases.
According to current genome assembly and ENCODE data housed at the UCSC Genome Browser, the so-called fusion site is located inside the first intron of the DDX11L2 gene on human chromosome 2. Furthermore, the putative fusion site has clear epigenetic and biochemical evidence of being a functional transcription factor binding motif, and being involved in the alternative transcriptional regulation of DDX11L2. There are actually three clear primary areas of transcription factor binding in DDX11L2, the first is in the promoter region directly adjacent to the first exon and the second is in the first intron corresponding to the fusion site sequence.
So why are multiple transcription factor binding regions often found in genes, as is the case with DDX11L2? A recent research paper has elucidated the complex nature of promoter-like regions in genes, showing that interacting promoters in genes possess combinatorial regulatory functions (Li et al. 2012). This point is the most pertinent in regards to the possible role that the putative fusion-like sequence is playing in the expression of the DDX11L2 gene. Given that transcripts of different lengths are produced from the DDX11L2 gene, along with the presence of a promoter-like region in the first intron, it is possible that this configuration is involved in the alternative splicing and regulation of the gene’s variable length products. It may also play some regulatory role via higher order chromatin structure in the expression of the adjacent WASH gene which overlaps with DDX11L2 by several base pairs, but is expressed in the plus strand (opposite) orientation (Costa et al. 2009).
The DDX11L2 gene is transcribed on the minus strand in the telomere to centromere orientation, so the fusion site sequence is actually interpreted by the transcriptional machinery in the reverse complement. Thus, the fusion sequence is not the standard plus strand sequence we are always given in the literature as evidence for a fusion—it is read in the reverse complement as part of a functional motif in a complexly transcribed gene. The gene which encompasses the fusion-like motif encodes at least two different consensus transcripts, and the sequences are associated with the RNA helicase family of genes.
Multiple transcriptionally active histone marks encompass and surround the fusion-like motif along with evidence for active open chromatin. Furthermore, significant levels of transcripts for the DDX11L2 gene have been characterized in at least 255 different cell lines and/or tissues. Coexpression for the DDX11L2 gene occurs with other genes associated with blood cell development and cell surface receptor activity associated with the extracellular matrix. It is also likely that the DDX11L2 gene regulates other genes for which it shares homology, such as the protein coding DDX11 gene that is coexpressed with DDX11L2. In addition, DDX11L2 transcript variants are characterized by a variety of miRNA binding sites as shown in this study, two of which are shared with the DDX11 protein coding gene. These data fit well with the emerging paradigm of pseudogene transcripts as key components of competitive endogenous RNA (ceRNA) networks that involve both miRNAs and protein coding transcripts (Ala et al. 2013; Taulli, Loretelli, and Pandolfi 2013). These complex sense and anti-sense interactions involve a complex regulatory network of cross-talk and competitive binding that is only beginning to be understood.
Costa et al. (2009), reported that at least 18 different DDX11L-like genes exist in the human genome. They also reported that very little synteny existed for these genes in apes. Using fluorescent in situ hybridization in chimpanzees and gorillas, only two locations of similarity for DDX11L-like genes were found in chimpanzee and four in gorilla—none of which corresponded to locations in human, or each other in apes. Of key importance to the topic of this paper was the fact that none of the regions of DDX11L hybridization in the chimp or gorilla genomes occurred on chromosomes 2A or 2B.
Other than the fact that the DDX11L2 gene is functional and is some sort of an RNA helicase regulatory ncRNA gene, little is known about its function. In fact, little is known about the majority of pseudogene-like sequences in the human genome, although data is beginning to rapidly accumulate showing that they are key regulators of transcription and translation, often in association with protein coding genes for which they share homology (Taulli, Loretelli, and Pandolfi 2013; Wen et al. 2012). The DDX11L2 gene would fall under the category of being an unprocessed pseudogene, meaning that it contains the standard intron-exon structure of protein coding genes along with a promoter region.
An unprocessed human pseudogene that has been well-studied is the PTEN pseudogene (PTENpg) which functions as part of a highly complex gene regulatory network (Johnsson et al. 2013). The PTENpg pseudogene encodes at least two different variants of regulatory RNA transcripts as part of an alternatively transcribed 4-exon gene.The two PTENpg RNAs regulate the transcripts of the protein coding PTEN gene by binding to them in a complementary fashion. Therefore, it is possible that the DDX11L2 gene may help regulate its protein coding homolog DDX11 as it is highly coexpressed with it in the same tissues and contains two similar miRNA binding sites indicating possible co-regulation via miRNAs. However, the DDX11L2 gene is also highly expressed with a large number of genes and is directly linked in extremely high levels of coexpression to three specific genes involved in blood cell development and chromatin remodeling. The other broad array of genes coexpressed with DDX11L2, but less directly connected are also largely involved in related functions and processes, including cell signaling in the extracellular matrix—a process intimately connected with blood cell/platelet generation and activity.
Functional, alternatively transcribed, post-transcriptionally spliced, post-transcriptionally regulated and network connected expressed genes, such as DDX11L2, cannot arise by the head-to-head fusion of chromosomes. Given the fact that all documented mammalian fusions in living animals only involve satDNA-satDNA or satDNA-telomereDNA fusions (Adega, Guedes-Pinto, and Chaves 2009; Tsipouri et al. 2008), the fusion-site negating data for human chromosome 2 then makes perfect sense. Clearly, the putative 800 base fusion site is not a degenerate fusion sequence, but a transcriptionally functional and active DNA binding motif read on the minus strand inside the DDX11L2 gene. Confirming this observation is the fact that there is no synteny in this region of human chromosome 2 for at least 614,000 bases encompassing the so-called fusion sequence compared to chimpanzee chromosomes 2A or 2B (Fan et al. 2002a; 2002b).
Evidence for macro-synteny outside the 615 Kb region surrounding the fusion site exists based on chromosomal banding (Yunis and Prakash 1982). This level of macro-synteny can be explained by the fact that gene order in the genome has been shown to be directly linked to categorical groups of function and transcription in diverse eukaryotes (Lopez, Guerra, and Samuelsson 2010). In fact, a sequential comparison of chimpanzee chromosomes 2A and 2B with human chromosome two in 300 base increments (irrespective of the linear order of fragments), revealed an overall DNA sequence similarity of 69% (Tomkins 2013). Since humans and apes do share high levels of macro-synteny among genes and chromosomes because biochemical function and transcription depend on it, this is to be expected. Of course, given the fact that the chimpanzee genome is primarily assembled based upon the human genomic framework, we really don’t know for sure how accurate the chimp genome assembly is at this point since it does not stand on its own merits (Tomkins 2011).
Combined with the fact that no valid evidence exists for a fossil centromere on human chromosome 2, the evolutionary idea of the chromosome two fusion in humans should be completely abandoned.
References
Abdelhaleem, M., L. Maltais, and H. Wain. 2003. The human DDX and DHX gene families of putative RNA helicases. Genomics 81, no. 6:618–622.
Adega, F., H. Guedes-Pinto, and R. Chaves. 2009. Satellite DNA in the karyotype evolution of domestic animals—clinical considerations. Cytogenetic and Genome Research 126, nos. 1–2:12–20.
Ala, U., F. A. Karreth, C. Bosia, A. Pagnani, R. Taulli, V. Léopold, Y. Tay, P. Provero, R. Zecchina, and P. P. Pandolfi. 2013. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proceedings of the National Academy of Sciences of the United States of America 110, no. 18:7154–7159.
Chang T-H., H-Y. Huang, J. B-K. Hsu, S-L. Weng, J-T. Horng, and H-D. Huang. 2013. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinformatics 14, Suppl 2:S4.
Chaves, R., F. Adega, J. Wienberg, H. Guedes-Pinto, and J. S. Heslop-Harrison. 2003. Molecular cytogenetic analysis and centromeric satellite organization of a novel 8;11 translocation in sheep: A possible intermediate in biarmed chromosome evolution. Mammalian Genome 14, no. 10: 706–710.
Cordin, O., J. Banroques, N. K. Tanner, and P. Linder. 2006. The DEAD-box protein family of RNA helicases. Gene 367:17–37.
Costa, V., A. Casamassimi, R. Roberto, F. Gianfrancesco, M. R. Matarazzo, M. D’Urso, M. D’Esposito, M. Rocchi, and A. Ciccodicola. 2009. DDX11L: A novel transcript family emerging from human subtelomeric regions. BMC Genomics 10:250.
Creyghton, M. P., A. W. Cheng, G. G. Welstead, T. Kooistra, B. W. Carey, E. J. Steine, J. Hanna, M. A. Lodato, G. M. Frampton, P. A. Sharp et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America 107, no. 50:21931–21936.
Dunham, I., A. Kundaje, S. F. Aldred, P. J. Collins, C. A. Davis, F. Doyle, C. B. Epstein, S. Frietze, J. Harrow, R. Kaul et al. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489, no. 7414:57–74.
Fairbanks, D. J. 2010. Relics of Eden: The powerful evidence of evolution in human DNA. Amherst, New York: Prometheus Books.
Fan, Y., E. Linardopoulou, C. Friedman, E. Williams, and B. J. Trask. 2002a. Genomic structure and evolution of the ancestral chromosome fusion site in 2q13-2q14.1 and paralogous regions on other human chromosomes. Genome Research 12, no. 11:1651–1662.
Fan, Y., T. Newman, E. Linardopoulou, and B. J. Trask. 2002b. Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions. Genome Research 12, no. 11:1663–1672.
Gerring, S. L., F. Spencer, and P. Hieter. 1990. The CHL 1 (CTF 1) gene product of Saccharomyces cerevisiae is important for chromosome transmission and normal cell cycle progression in G2/M. The EMBO Journal 9, no. 13:4347–4358.
Harmston, N. and B. Lenhard. 2013. Chromatin and epigenetic features of long-range gene regulation. Nucleic Acids Research 14, no. 15:7185–7199.
Hruz, T., O. Laule, G. Szabo, F. Wessendorp, S. Bleuler, L. Oertle, P. Widmayer, W. Gruissem, and P. Zimmermann. 2008. Genevestigator v3: A reference expression database for the meta-analysis of transcriptomes. Advances in Bioinformatics 2008: 420747, doi:10.1155/2008/420747.
Ijdo, J. W., A. Baldini, D. C. Ward, S. T. Reeders and R. A. Wells. 1991. Origin of human chromosome 2: An ancestral telomere-telomere fusion. Proceedings of the National Academy of Sciences of the United States of America 88, no. 20:9051–9055.
Johnsson, P., A. Ackley, L. Vidarsdottir, W. O. Lui, M. Corcoran, D. Grander, and K. V. Morris. 2013. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nature Structural & Molecular Biology 20, no. 4:440–446.
Li, G., X. Ruan, R. K. Auerbach, K. S. Sandhu, M. Zheng, P. Wang, H. M. Poh, Y. Goh, J. Lim, J. Zhang et al. 2012. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, no. 1:84–98.
Lopez, M D., J. J. M. Guerra, and T. Samuelsson. 2010. Analysis of gene order conservation in eukaryotes identifies transcriptionally and functionally linked genes. PloS ONE 5, no. 5:e10654, doi:10.1371/journal.pone.0010654
Obayashi, T., Y. Okamura, S. Ito, S. Tadaka, I. N. Motoike, and K. Kinoshita. 2013. COXPRESdb: A database of comparative gene coexpression networks of eleven species for mammals. Nucleic Acids Research 41(Database issue): D1014-20, doi: 10.1093/nar/gks1014.
Pasquinelli, A. E. 2012. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nature Reviews Genetics 13, no. 4:271–282.
Salmena, L., L. Poliseno, Y. Tay, L. Kats, and P. P. Pandolfi. 2011. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 146, no. 3:353–358.
Taulli, R., C. Loretelli, and P. P. Pandolfi. 2013. From pseudoceRNAs to circ-ceRNAs: A tale of cross-talk and competition. Nature Structural & Molecular Biology 20, no. 5:541–543.
Tomkins, J. P. 2011. How genomes are sequenced and why it matters: Implications for studies in comparative genomics of humans and chimpanzees. Answers Research Journal 4:81–88. Retrieved from http://www.answersingenesis.org/articles/arj/v4/n1/implications-for-comparative-genomics.
Tomkins, J. P. 2013. Comprehensive analysis of chimpanzee and human chromosomes reveals average DNA similarity of 70%. Answers Research Journal 6:63–69. Retrieved from http://www.answersingenesis.org/articles/arj/v6/n1/human-chimp-chromosome.
Tomkins, J. P. and J. Bergman. 2011a. The chromosome 2 fusion model of human evolution—part 2: Re-analysis of the genomic data. Journal of Creation 25, no. 2:111–117.
Tomkins, J. P. and J. Bergman. 2011b. Telomeres: Implications for aging and evidence for intelligent design. Journal of Creation 25, no. 1:86–97.
Tsipouri, V., M. G. Schueler, S. Hu, NIS Comparative Sequencing Program, A. Dutra, E. Pak, H. Riethman, and E. D. Green. 2008. Comparative sequence analyses reveal sites of ancestral chromosomal fusions in the Indian muntjac genome. Genome Biology 9, no. 10:R155, doi:10.1186/gb-2008-9-10-r155.
Ventura, M., C. R. Catacchio, S. Sajjadian, L. Vives, P. H. Sudmant, T. Marques-Bonet, T. A. Graves, R. K. Wilson, and E. E. Eichler. 2012. The evolution of African great ape subtelomeric heterochromatin and the fusion of human chromosome 2. Genome Research 22, no. 6:1036–1049.
Wen, Y. Z., L. L. Zheng, L. H. Qu, F. J. Ayala, and Z. R. Lun. 2012. Pseudogenes are not pseudo any more. RNA Biology 9, no. 1:27–32.
Wu, C., I. Macleod, and A. Su. 2013. BioGPS and MyGene.info: Organizing online, gene-centric information. Nucleic Acids Research 41 (Database issue):D561-565, doi: 10.1093/nar/gks1114.
Yunis, J. J. and O. Prakash. 1982. The origin of man: A chromosomal pictorial legacy. Science 215, no. 4539: 1525–1530.
Zentner, G. E., P. J. Tesar, and P. C. Scacheri. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Research 21, no. 8: 1273–1283.






