The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
One of the iconic (yet enigmatic) arguments for human-ape common ancestry has been the β-globin pseudogene (HBBP1). Evolutionists originally speculated that apparent mutations in HBBP1 were shared mutational mistakes derived from a human-chimpanzee common ancestor. However, others noted that if the gene was indeed non-functional, then it should have mutated markedly in the past 3 to 6 million years of human evolution due to a lack of selective constraint on the region. Recent research confirms that the HBBP1 region of the 6-gene β-globulin cluster is highly non-variable compared to the other β-globin genes based on large-scale DNA diversity assessment within both humans and chimpanzees. Highlighting the lack of HBBP1 sequence variability is genetic data from three different reports that link point mutations in the HBBP1 gene with β-thalassemia disease pathologies. Biochemical evidence for functionality is indicated by multiple categories of functional genomics data showing that the HBBP1 gene is transcriptionally active and a key interactive component of the β-globin gene network. In brief, the HBBP1 gene encodes two consensus regulatory RNAs that are alternatively transcribed and/or post-transcriptionally spliced. This functional complexity produces at least 16 different exon variant transcripts and 42 different intron variant transcripts. Two major regulatory regions in the HBBP1 locus contain active transcription factor binding sites that overlap multiple categorical regions of epigenetic data for functionally active chromatin. The HBBP1 gene also has the most regulatory associations with active and open chromatin within the entire β-globin cluster and its transcripts are expressed in at least 251 different human cell and/or tissue types. Instead of being a useless genomic fossil according to evolutionary predictions, the HBBP1 gene appears to be a highly functional and cleverly integrated feature of the human genome that is intolerant of mutation.
Keywords: β-globin pseudogene, HBBP1, human evolution, ENCODE
Introduction
Hemoglobin is a protein in red blood cells that transports oxygen throughout the body in the circulatory system. The human hemoglobin protein consists of a cluster of two chains of different subunit proteins. One of these chains is called the α-globins, which remain the same over the course of embryo development to adulthood (Bank 2006; Sankaran, Xu, and Orkin 2010). The second set is called the β chain, which specifically changes in subunit composition at the embryo-to-fetal transition and the fetal-to-adult transition during human development. Remarkably, the subsets of globin genes switch on and off during development in the same linear order in which they are located on the chromosome (Bank 2006; Sankaran, Xu, and Orkin 2010). This amazing bioengineering within the β-globin locus allows the developing embryo and infant to receive oxygen at the correct levels in its system throughout its critical growth processes and developmental transitions.
The β-globin proteins are encoded in a cluster of six genes: HBE1, HBG2, HBG1, HBBP1, HBD, and HBB (see fig. 1) surrounded by a large complex of class I olfactory receptor genes (Molete et al. 2002, Sheffield et al. 2013). The β-globin genes are encoded on the reverse (minus) strand and transcribed 5′ to 3′ in the centromere-to-telomere orientation. The β-globin cluster extends over 80,000 bases on chromosome 11 and is known collectively as HBB. The embryonic-to-adult growth stage expression of each gene in the cluster depends on each gene’s long-range interaction with a control region containing multiple sites of active open chromatin (DNase hypersensitive sites) preceding the cluster (6 to 18 Kb) called the “locus control region” or LCR (Dean 2011; Molete et al. 2002; Sheffield et al. 2013). While five out of the six genes in the β-globin cluster encode functional proteins, one of the genes called HBBP1 does not produce a recognizable protein product. An early comparative analysis of the HBBP1 gene in humans, chimpanzees, and gorillas indicated that each taxon shared the same inferred mutations (substitutions) that negated the production (translation) of a functional protein. These inferred anomalies were in the initiator codon, a substitution in codon 15, which generates a termination signal, and a nucleotide deletion in codon 20, resulting in a frame shift which yields a variety of termination signals in exons 2 and 3 (Chang and Slightom 1984).
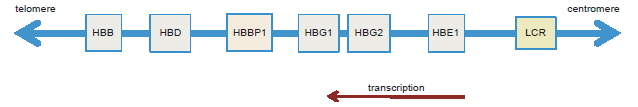
Fig. 1. Graphical representation of the β-globin gene cluster showing the arrangement of the five protein coding genes, the HBBP1 pseudogene, and the locus control region (LCR).
Outside of these presumed code errors, HBBP1 contains all the hallmarks of a functional gene including an intact promoter and clear exon-intron junctions. This would place it in the category of being an “unprocessed pseudogene.” In the general evolutionary model, these types of sequences are thought to have arisen primarily from a duplication event and are often annotated as being related to a specific “parent gene” from which they are presumed to have been derived (See recent review of pseudogenes by Wen et al. 2012).
Evolutionists have also assumed that because the same alleged HBBP1 gene mistakes were found in both humans and apes, that this was clear evidence of common ancestry. Several popular evolutionary science writers have capitalized on this idea claiming that the preliminary evidence of non-function along with sequence similarity was clear proof of common ancestry between humans and the great apes. The general idea is that the HBBP1 pseudogene is a genomic record of shared mistakes passed along over deep evolutionary time. Most notable among the efforts promoting this idea are excerpts from books written by Miller (2009, pp. 101–112) and Fairbanks (2010, pp. 56–57). Of course, the hypothesis of the HBBP1 gene being an evolutionary genomic fossil largely hinges upon the non-functionality of the sequence as claimed by both Miller and Fairbanks. However, multiple lines of evidence from recently reported research and publicly available data sets now indicate that the HBBP1 is functional and critical to healthy blood chemistry, thus negating the arguments promoted by Miller and Fairbanks.
Human-Ape HBBP1 Similarity
Since recent data regarding the exact sequence similarities between the human HBBP1 gene locus and various apes was not found, a current assessment of the sequence identity was performed using standard web tools available at the UCSC Genome Browser (public DNA sequence database with interactive web tools; genome.ucsc.edu).
Based on current data in the UCSC genome browser for human (HRCh37/hg19), the HBBP1 locus is located on chromosome 11 (region p15.4) and comprises a contiguous sequence of about 2,350 bases. A comparative analysis of the human HBBP1 locus with the chimpanzee (panTro4), gorilla (goGor3.1), orangutan (ponAbe2), and macaque (rheMac3) genomes, indicates that overall it is 97.8%, 97.8%, 92.2%, and 92.1% identical, respectively (including insertions and deletions).
Clearly, the HBBP1 gene is highly similar between humans and various primates, but not completely identical. Both creationists and evolutionists would generally agree that this sequence conservation between taxa indicates functionality. Evolutionists would maintain that the functionality of the sequence constrained selection over long periods of time, yet derived in humans via descent from a common ancestor with apes. Creationists would postulate that the sequence similarities were a matter of code re-use for a common functional purpose, a theme inherent to designed and engineered systems. However, orthologous gene networks with similar functions across disparate taxa may be composed of non-identical sets of genes (Al-Shahrour et al. 2010). A feature not explainable by gradualistic models of evolution, but common to complex engineered systems. As we shall see, HBBP1 is highly functional with a wide variety of transcripts, genomic network connections, epigenetic annotations, and tissue/cell type expression profiles. But first, it’s important to briefly review an early report and recent research linking functionality with HBBP1 sequence conservation (non-variability).
HBBP1 is Non-Variable and Mutation Intolerant
One of the main problems with the “shared mistakes” and common ancestry application to the HBBP1 data is that the actual evidence for the claim is contradictory to theoretical evolutionary processes of selection and sequence divergence. If HBBP1 is a defunct non-functional sequence, mutations should have freely accumulated within it creating significantly more divergence. In fact, Chang and Slightom in their original paper noted that
The three hominoid psi beta-globin genes [betaglobin pseudogenes] show a high degree of sequence correspondence, with the number of differences found among them being only about one-third of that predicted for DNA sites evolving at the neutral rate (i.e. for sites evolving in the absence of purifying selection). (Chang and Slightom 1984, p. 767)
A recently published research report revisited this issue of extreme conservation in the HBBP1 gene among humans and chimpanzees using bioinformatics (Moleirinho et al. 2013). The researchers compared the entire β-globin gene cluster in over 1,000 human individuals using data from the “1,000 Genomes Project.” They also assessed the diversity of the β-globin gene cluster among a large number of chimpanzees using data from the “PanMap Project.” Out of the six genes in the β-globin cluster, the HBD gene and its linked neighbor, the HBBP1 pseudogene, exhibited much less genetic diversity compared to the other β-globin genes in both humans and chimpanzees. The researchers concluded that “[comprehensive] analyses, based on classic neutrality tests, empirical and haplotype-based studies, revealed that HBD and its neighbor pseudogene HBBP1 have mainly evolved under purifying selection, suggesting that their roles are essential and nonredundant.” Within the evolutionary model, non-variability and conservation of sequence are interpreted as constraint on genomic sequences and regarded as an indicator of biological function (Svensson, Arvestad, and Lagergren 2006).
Another important piece of data indicating functionality for the HBBP1 pseudogene is supported by three different recent research reports that studied the genetics of human blood pathology in relation to the genes in the β-globin cluster (Giannopoulou et al. 2012; Nuinoon et al. 2010; Roy et al. 2012). In these studies, researchers showed that a variety of single point mutations in the HBBP1 pseudogene were closely associated with variations (mild to severe) of a human blood disease called β-thalassemia. This is a disease condition associated with alterations of the β-globin gene cluster which results in the persistence of fetal hemoglobin past the fetal stages into early childhood and adulthood (Rees et al. 1999). In addition to the β-thalassemia connection, the UCSC genome browser indicates that the HBBP1 region is a quantitative trait locus (QTL, see glossary) associated with osteoarthritis. These genetic data not only add additional support to the putative functional nature of the gene, but also emphasize the fact that very little genetic variation is tolerated in the locus as described by Moleirinho et al. (2013).
The HBBP1 Pseudogene Is Functional
Not only did Moleirinho et al. (2013) determine that the HBBP1 pseudogene was markedly non-variable and likely functional, they verified the hypothesis of inferred functionality with data from the ENCODE project (Dunham et al. 2012). Moleirinho et al. detected significant interactions between a segment of the β-globin cluster comprising both HBD and HBBP1, and different regions upstream of the lead gene HBE, which overlap the locus control region (LCR). As mentioned earlier, the LCR is the main control region approximately 6,000 to 18,000 bases from the β-globin cluster that engages in long range interactions via complex chromatin loops with the globin genes in the cluster (Dean 2011; Xu et al. 2010; Xu et al. 2012). Moleirinho et al. also elaborated that
both HBD and HBBP1 may act as anchor regions in LCR-driven chromatin looping, a crucial mechanism for temporal coordination of gene expression in the human beta-globin cluster (Moleirinho et al. 2013, p. 567).
A variety of previous papers have documented complex transcriptional control combined with long range chromatin interactions in the β-globin locus (Deng et al. 2012; Dostie et al. 2006; Xu et al. 2010).
These observations by Moleirinho et al. are further validated and augmented by yet another recent report by Sheffield et al. (2013) in which the HBBP1 pseudogene was shown to have at least eight network correlations within a wide variety of open and active transcriptional control sites across the β-globin locus. This is more than any other gene in the β-globin cluster. These data were derived from hematopoietic (blood) stem cells. The integrated data was based on the analysis of open and active chromatin determined by DNase1 sensitivity—a highly accurate regulatory indicator of functional chromatin (Thurman et al. 2012). The DNase results were also correlated with large-scale gene expression data.
Additional proof of gene function is the identification and characterization of a transcriptional product(s). In the case of the HBBP1 pseudogene, its multiple gene products are regulatory RNAs. In the UCSC genome browser ENCODE version 14 comprehensive gene annotation set for the HBBP1 pseudogene, annotation tracks are shown for the localization of 14 spliced expressed sequences (chr11: 5263100-5265425) that align within the HBBP1 locus as shown in Fig. 2. Many of these represent processed transcripts and/or the products of alternative splicing/transcription. The Ensembl database lists two main consensus (reference) transcripts (ENST00000433329, ENST00000454892) of 439 and 455 bases in length. One consensus version contains two exons while the other has three, and there are alternatively spliced variants of these as shown in the Vega Genome Browser (ensembl.org) for the manually curated “Havana” annotations for HBBP1. The Ensembl gene variation data set for HBBP1 lists 16 different exon variant transcripts and 42 different intron variant transcripts (useast.ensembl.org/Homo_sapiens/Gene/Variation_Gene/Table?g=ENSG00000229988;r=11: 5263184-5264767). This diversity of transcript variation is partially facilitated by six different sets of exon start/end sites within the HBBP1 gene as described at the GeneLoc database at the Weizmann Institute (genecards.weizmann.ac.il/geneloc). These expressed sequence regions in the HBBP1 gene overlap and correspond with annotation tracks for transcriptionally active chromatin and transcription factor binding discussed in more detail below (see fig. 2).
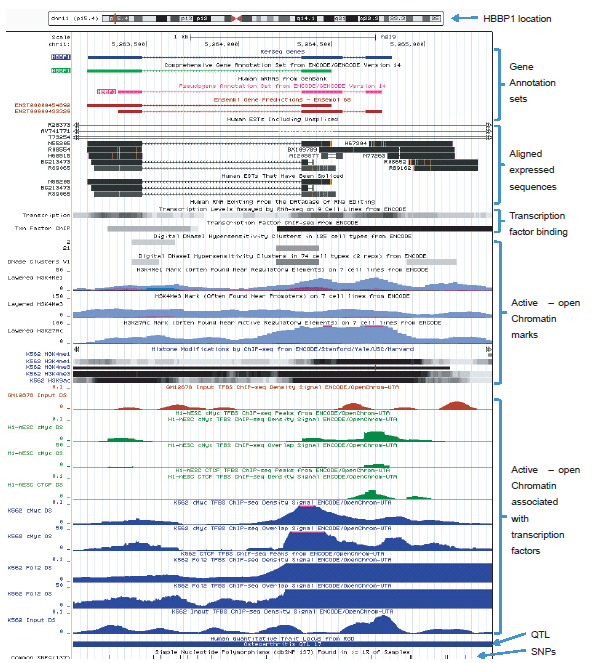
Fig. 2. UCSC genome browser data showing selected gene annotation and ENCODE-related tracks for the HBBP1 locus. Analysis image accessed at genome.ucsc.edu on May 7, 2013. View larger image.
A breakdown of the transcriptional profiles for a wide variety of human pseudogenes is also located at the pseudoMap database (Chan et al. 2013a; pseudomap.mbc.nctu.edu.tw). The current entry for the HBBP1 pseudogene lists the two ENSEMBL consensus IDs and indicates that the target gene regulated by the two HBBP1 transcripts is HBE1, the assumed parent (via hypothetical gene duplication) and the first gene in the β-globin cluster. However, the recent data published by Sheffield et al. (2013) clearly shows that the regulatory activity of the HBBP1 pseudogene associates across a wide variety of functional chromatin sites in the β-globin cluster.
Database Survey of HBBP1 Tissue/Cell Expression Profiles
Sheffield et al. (2013) also announced the formation of a new online database that listed much of the unpublished data not discussed in their paper called the “Regulatory Elements Database” (dnase.genome.duke.edu). Interestingly, 74 different DNase hypersensitive network site connections are listed for the HBBP1 gene. DNase hypersensitive sites are open/active areas of chromatin associated with long-range interactions and gene regulation/transcription (Thurman et al. 2012). The Regulatory Elements Database entry for HBBP1 also lists gene expression profiles that show transcriptional activity for HBBP1 in brain, endothelial, epithelial, fibroblast, hematopoietic, liver, muscle, and stem cells.
The BioGPS (biogps.org) gene annotation and analysis portal lists significant levels of expression data for the HBBP1 gene in 84 different specific tissue and cell types. This translates into the HBBP1 gene being expressed in 28 out of 31 major categorical tissue groupings in the human body as summarized in the HBBP1 gene entry in the GeneCards human gene compendium (www.genecards.org) and shown in Fig. 3. Interestingly, an even more thorough compilation of HBBP1 gene function is available at the GeneVestigator web portal (www.genevestigator.com) which shows positive levels of expression for HBBP1 across 251 different tissue and cell types with normalized comparisons to other genes of similar expression intensity. For obvious reasons, this data was too extensive to show in a figure in this report, but can be readily accessed using “hbbp1” as the search query at genevestigator.com. For a summary of the database survey results, see Table 1.
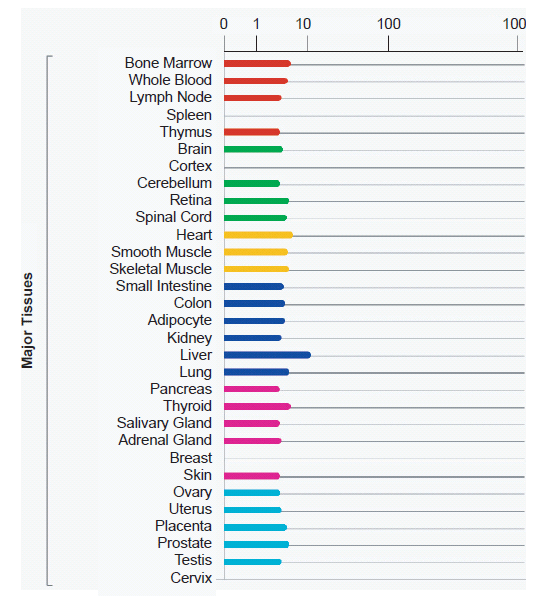
Fig. 3. Human tissue-based summary of HBBP1 pseudogene expression compiled from data across 84 different tissues/cell lines (BioGPS.org). Analysis image accessed from genecards.org on May 7, 2013.
| Number of tissues/cell types expression detected | Database URL |
| 8 | dnase.genome.duke.edu |
| 28 | www.genecards.org |
| 84 | Biogps.org |
| 251 | www.genevestigator.com |
ENCODE Data Mining for the HBBP1 Gene
According to the most recent ENCODE release of data at the UCSC Genome Browser (Pei et al. 2012; Rosenbloom et al. 2013; genome.ucsc.edu) for the human HBBP1 locus (chr11: 5263100-5265425), multiple overlapping annotation tracks are listed for the following functional genomic features which are also graphically depicted in Fig. 2:
- Consensus/reference sequences for the two main transcript classes encoded by the HBBP1 locus as depicted by ENCODE/GENCODE version 14 annotations, NCBI reference sequence, and Ensembl gene predictions. Validating these reference sequences is a wide variety of aligned expressed transcript sequences. In addition, there are a variety of aligned expressed sequences that are not represented in the consensus gene annotations.
- Several clearly distinguished tracks showing functional transcription factor binding domains based on binding site sequence homology and coprecipitated DNA-protein complexes (ChIP-seq data; see glossary). Multiple transcription factors, regulatory proteins, and chromatin loops have been characterized at the β-globin locus in long range chromatin regulatory interactions (Deng et al. 2012; Holwerda and de Laat 2012; Xu et al. 2010).
- Multiple clearly distinguished tracts of functionally active/open chromatin based on multiple annotation layers of DNase hypersensitive sites and combinatorial histone marks associated with active gene transcription that functionally overlap (see glossary). As mentioned above, the work by Sheffield et al. (2013) further elaborates and defines the open chromatin landscape at HBBP1 and associates it specifically with gene expression data. Of particular importance is the extensive combinatorial presence of acetylation (H3K27ac, H3K9ac) and methylation (H3K4Me1, H3K4Me3) histone marks (peaks) across the entire HBBP1 region that unequivocally distinguishes the region as transcriptionally active. The H3K27ac histone acetylation marks are definitively associated with active enhancer elements in long-range chromatin interactions associated with transcription (Creyghton et al. 2010; Zentner, Tesar, and Scacheri 2011) and are also associated with active gene promoters (Dunham et al. 2012). The H3K9ac histone acetylation marks combined with the H3K4Me1 and H3K4Me3 histone methylation signatures are also clearly indicative of active gene promoters and transcription (Dunham et al. 2012; Hon, Hawkins, and Ren 2009; Wang et al. 2008).
- Active and open areas of chromatin physically associated with a variety of canonical transcription-related DNA-binding proteins. The fact that transcriptionally defined epigenetic marks overlap with a variety of known and bound transcription factors coupled with a wide variety of annotated transcripts, is exceptionally strong support for functionality at this locus.
- A QTL for osteoarthritis (see glossary for QTL definition). Not shown in the UCSC annotation tracks are the associations between point mutations in the HBBP1 gene and various pathologies of the blood disease β-thalassemia (Giannopoulou et al. 2012; Nuinoon et al. 2010; Roy et al. 2012). These genetic associations at the HBBP1 locus with human health illustrate the importance of the gene for human development and physiology. These genetic findings also mesh well with the wide variety of ENCODE and gene annotation tracks to more clearly highlight the functional importance of HBBP1.
- Single nucleotide polymorphisms (SNPs) occurring predominantly in non-exonic regions of the HBBP1 locus. The locations of these SNPs within the HBBP1 locus emphasize the non-variable and mutation-intolerant nature of the locus, particularly in its key RNA coding regions.
Summary and Conclusion
One of the key emerging concepts of the molecular biology revolution is that the concept of what defines a “gene” is becoming elusive and difficult to define due to the extreme complexity of the genome (reviewed by Portin 2009). This is particularly true when it comes to the concept of pseudogenes. Pseudogenes are typically similar to other genes in the genome except that they have apparent coding-sequence deficiencies like frameshifts and premature stop codons. For a definition of pseudogene types, see the glossary at the end of this paper.
Pseudogenes are ubiquitous and abundant in plant and animal genomes and in the past have been referred to as “genomic fossils” and considered to be “junk DNA.” However, it has been proved that an increasing number of pseudogenes play essential roles in the regulation of other genes and are transcribed into a variety of functional RNAs ( Li, Yang, and Wang 2013; Tutar 2012; Wen et al. 2012). Pseudogene RNA transcripts can be processed into small interfering RNA molecules that bind to and regulate full-length transcripts or they can regulate and fine-tune the presence of another class of small transcriptional regulators called micro RNAs (Li, Yang, and Wang 2013; Tutar 2012). Pseudogenes have also been found to be key regulators of human health in regard to their control of oncogenes and other cell cycle regulatory genes (Chan et al. 2013b; Tutar 2012; Wen et al. 2012). The clear fact is that our current knowledge of these types of genes remains poorly understood and their pseudo status appears to be rapidly diminishing the more that we discover about them.
In regards to hypothetical ideas about human evolution, one of the most commonly used arguments for “shared mistakes” inherited supposedly through common descent between humans and great apes has been the HBBP1 pseudogene. On the other hand, it is also one of the most perplexing arguments for human evolution because the sequence is so well conserved among humans and apes. According to evolutionary dogma, selective restraints should have been lifted on the pseudogene gene millions of years ago, thus allowing it to mutate freely.
The non-variable enigmatic nature of the HBBP1 locus originally noticed by Chang and Slightom (1984) was but a foreshadowing of its true highly functional nature. The assessed HBBP1 sequence diversity within both humans and chimpanzees verified that the region was highly non-variable compared to other genes in the β-globin cluster using extensive modern DNA variation databases—a hallmark of functionally important DNA sequence that serves a common purpose across taxa (Moleirinho et al. 2013). Highlighting the importance of the genetic nondiversity data are three recent discoveries showing that single base mutations in the HBBP1 pseudogene are associated with various pathologies of a blood disease called β-thalassemia (Giannopoulou et al. 2012; Nuinoon et al. 2010; Roy et al. 2012).
The inferred functionality based on non-variability and genetic disease association studies have been spectacularly vindicated by multiple biochemical categories of ENCODE data showing that the HBBP1 pseudogene encodes two consensus regulatory RNAs along with multiple other transcript variants due to alternative splicing and parent transcript processing into smaller regulatory sequences (Moleirinho et al. 2013; Sheffield et al. 2013; UCSC Genome Browser ENCODE v14 tracks). The HBBP1 pseudogene also has the most regulatory associations within the β-globin cluster for DNase hypersensitive sites and its transcripts are expressed in at least 251 different tissues and cell types (dnase.genome.duke.edu, BioGPS.org, Genevestigator.com).
Other types of functional ENCODE data include the identification of transcription factor binding in several key regulatory regions of the HBBP1 gene. These active areas of transcription are also characterized by a variety of histone methylation and acetylation marks commonly identified with active regulatory regions associated with both transcription and/or long-range chromatin interactions. Open and active chromatin that overlaps with the other epigenetic marks is also found throughout the HBBP1 gene as identified by sectors of DNase hypersensitivity. Taken together, these combinatorial lines of both genetic and functional biochemical evidence strongly indicate that the HBBP1 gene is anything but pseudo.
Instead of being a useless mutated remnant according to failed evolutionary predictions, the HBBP1 β-globin pseudogene appears to be playing multiple key functional roles in a wide diversity of tissues and cell types as a cleverly engineered regulatory feature that is highly intolerant of mutation.
Glossary of Terms
Alternative Splicing: The process of producing a wide variety of messenger RNA transcripts from a single gene (Barash et al. 2010). Most human genes produce a wide variety of transcript variants from alternative transcription start sites and from the alternative post-transcriptional splicing of exons. Transcript variation is also affected by variable splice signals present in introns. See gene structure below for more information.
ChIP-sequencing (ChIP-seq): A method used to identify DNA protein binding sites and their physical interactions by combining chromatin immunoprecipitation (ChIP) with massively high-throughput DNA sequencing (Jothi et al. 2008; Landt et al. 2012).
Chromatin: The combination of DNA and the proteins it is associated and packaged with that make up the contents of chromosomes.
DNase sensitivity: DNases are a class of enzymes that cut open sections of DNA associated with active regions of the genome. DNase sensitivity assays use this property to identify and sequence active chromatin (Thurman et al. 2012).
ENCODE: As stated at the ENCODE-NIH web site, “The National Human Genome Research Institute (NHGRI) launched a public research consortium named ENCODE, the Encyclopedia Of DNA Elements, in September 2003, to carry out a project to identify all functional elements in the human genome sequence.” ENCODE is currently ongoing and for all practical purposes, has just begun to unravel the mystery of the genome in a limited number of cell types (Birney et al. 2007; Dunham et al. 2012).
Epigenetic modification: The combination of histone modification and methylation of DNA. See terms for “methylation” and “histone modification.” Also known as chromatin marks. Epigenetic profiles (marks) are a very accurate indicator of transcriptional and regulatory activity in the genome.
Gene structure: Genes in multicellular organisms are composed of coding and non-coding segments. The coding segments, called exons, code for the final protein product if the gene is translated. In many cases, the gene codes for an RNA molecule that is used for either structural or regulatory purposes. There are numerous categories of noncoding RNA. The intervening and non-coding parts of genes, are called introns which contain control sequences and features that facilitate transcription and splicing of the mRNA produced (transcribed) from a gene.
Histone modification (marking): The addition of chemical modifications (tags) on histones, the proteins that associate with and package DNA to help regulate varying levels of genetic activity and/ or repression. There are a wide variety of histone marks that can be assayed across the genome that are related to the covalent modification processes of acetylation and methylation of amino acids in histone N-terminal tails. The types and combinations of histone tags in various regions of the genome provide accurate indicators of genetic activity (Zentner, Tesar, and Scacheri 2011).
Long-range chromatin interactions: The formation of interactive loops of chromatin with enhancer sequences, transcription factors, and other DNA binding proteins. This 3-D based view of gene regulation is emerging as a key feature in how genes are regulated and expressed (Dean 2011; Deng et al. 2012; Holwerda and de Laat 2012).
Pseudogene: DNA features that were once thought to be nothing but genomic fossils representing defunct gene sequences, hence the name. There are two general types of pseudogenes based on their DNA signatures. One is called the “unprocessed” pseudogene that has all of the basic elements of a regular gene (promoter, exons, introns) but has alterations that prevent it from producing a viable protein product. The second type is called a “processed” pseudogene and thought to represent a mRNA that was reverse transcribed into DNA and then inserted into the genome. These types lack the exon-intron structure of standard genes. However, both types of pseudogenes are now being found to produce regulatory RNA molecules and be functionally important to human health. See recent review by Wen et al. (2012).
QTL (quantitative trait locus/loci): Locations in the genome that genetically associate with and contribute to the variability of quantitative (multigenic) complex traits.
Transcription factor binding: Transcription factors are DNA binding proteins that are associated with the active transcription of DNA—the copying of a genomic region to produce an RNA molecule (transcript) by DNA-dependent RNA polymerases. RNA transcripts can code for proteins or noncoding RNAs that are regulatory or structural in purpose (Landt et al. 2012; Lee et al. 2012).
References
Al-Shahrour, F., P. Minguez, T. Marqués-Bonet, E. Gazave, A. Navarro, and J. Dopazo. 2010. Selection upon genome architecture: Conservation of functional neighborhoods with changing genes. PLoS Computational Biology 6, no. 10: e1000953.
Bank, A. 2006. Regulation of human fetal hemoglobin: New players, new complexities. Blood 107, no. 2: 435–443.
Barash, Y., J. A. Calarco, W. Gao, Q. Pan, X. Wang, O. Shai, B. J. Blencowe, and B. J. Frey. 2010. Deciphering the splicing code. Nature 465, no. 7294: 53–59.
Birney, E., J. A. Stamatoyannopoulos, A. Dutta, R. Guigó, T. R. Gingeras, E. H. Margulies, Z. Weng, M. Snyder, E. T. Dermitzakis, et al. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816.
Chan, W.-L., W.-K. Yang, H.-D. Huang, and J.-G. Chang. 2013a. pseudoMap: An innovative and comprehensive resource for identification of siRNA-mediated mechanisms in human transcribed pseudogenes. Database 2013: bat 001. doi: 10.1093/database/bat001.
Chan, W.-L., C. Y Yuo, W. K. Yang, S. Y. Hung, Y. S. Chang, C. C. Chiu, K. T. Yeh, H. D. Huang, and J. G. Chang. 2013b. Transcribed pseudogene ψPPM1K generates endogenous siRNA to suppress oncogenic cell growth in hepatocellular carcinoma. Nucleic Acids Research 41, no. 6: 3734–3747.
Chang, L.-Y. E. and J. L. Slightom. 1984. Isolation and nucleotide sequence analysis of the β-type globin pseudogene from human, gorilla and chimpanzee. Journal of Molecular Biology, 180, no. 4: 767–783.
Creyghton, M. P., A. W. Cheng, G. G. Welstead, T. Kooistra, B. W. Carey, E. J. Steine, J. Hanna, M. A. Lodato, G. M. Frampton, P. A. Sharp, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America 107: 21931–21936.
Dean, A. 2011. In the loop: Long chromatin interactions and gene regulation. Briefings in Functional Genomics 10, no. 1: 3–10.
Deng, W., J. Lee, H. Wang, J. Miller, A. Reik, P. D. Gregory, A. Dean, and G. A. Blobel. 2012. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149, no. 6: 1233–1244.
Dostie, J., T. A. Richmond, R. A. Arnaout, R. R. Selzer, W. L. Lee, T. A. Honan, E. D. Rubio, et al. 2006. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Research 16, no. 10: 1299–1309.
Dunham, I., K. Beal, A. Brazma, P. Flicek, J. Herrero, N. Johnson, D. Keefe, et al. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489, no. 7414: 57–74.
Fairbanks, D. J. 2010. Relics of Eden: The powerful evidence of evolution in human DNA. Amherst, New York: Prometheus Books.
Giannopoulou, E., M. Bartsakoulia, C. Tafrali, A. Kourakli, K. Poulas, E. F. Stavrou, A. Papachatzopoulou, M. Georgitsi, and G. P. Patrinos. 2012. A single nucleotide polymorphism in the HBBP1 gene in the human β-globin locus is associated with a mild β-thalassemia disease phenotype. Hemoglobin 36, no. 5: 433–445.
Holwerda, S. and W. de Laat. 2012. Chromatin loops, gene positioning, and gene expression. Frontiers in Genetics 3: 217. doi: 10.3389/fgene.2012.00217.
Hon, G. C., R. D. Hawkins, and B. Ren. 2009. Predictive chromatin signatures in the mammalian genome. Human Molecular Genetics 18, no. R2: R195–R201.
Jothi, R., S. Cuddapah, A. Barski, K. Cui, and K. Zhao. 2008. Genome-wide identification of in vivo protein–DNA binding sites from ChIP-seq data. Nucleic Acids Research 36, no. 16: 5221–5231.
Landt, S. G., G. K. Marinov, A. Kundaje, P. Kheradpour, F. Pauli, S. Batzoglou, B. E. Bernstein, et al. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research 22, no. 9: 1813–1831.
Lee, B. K., A. A. Bhinge, A. Battenhouse, R. M. McDaniell, Z. Liu, L. Song, Y. Ni, et al. 2012. Cell-type specific and combinatorial usage of diverse transcription factors revealed by genome-wide binding studies in multiple human cells. Genome Research 22, no. 1: 9–24.
Li, W., W. Yang, and X-J. Wang. 2013. Pseudogenes: Pseudo or real functional elements? Journal of Genetics and Genomics 40, no. 4: 171–177.
Miller, K. R. 2009. Only a theory: Evolution and the battle for America’s soul. New York, New York: Penguin.
Moleirinho, A., S. Seixas, A. M. Lopes, C. Bento, M. J. Prata, and A. Amorim. 2013. Evolutionary constraints in the β-globin cluster: The signature of purifying selection at the δ-globin (HBD) locus and its role in developmental gene regulation. Genome Biology and Evolution 5, no. 3: 559–571.
Molete, J. M., H. Petrykowska, M. Sigg, W. Miller, and R. Hardison. 2002. Functional and binding studies of HS3.2 of the beta-globin locus control region. Gene 283, no. 1–2: 185–197.
Nuinoon, M., W. Makarasara, T. Mushiroda, I. Setiangingsih, P. A. Wahidiyat, O. Sripichai, N. Kumasaka, A. Takahashi, S. Svasti, T. Munkongdee, et al. 2010. A genome-wide association identified the common genetic variants influence disease severity in beta0-thalassemia/hemoglobin E. Human Genetics 127, no. 3: 303–314.
Pei, B., D. Sisu, A. Frankish, C. Howald, L. Habegger, X. J. Mu, R. Harte, et al. 2012. The GENCODE pseudogene resource. Genome Biology 13, no. 9: R51.
Portin, P. 2009. The elusive concept of the gene. Hereditas 146, no. 3: 112–117.
Rees, D. C., J. B. Porter, J. B. Clegg, and D. J. Weatherall. 1999. Why are hemoglobin F levels increased in HbE/β thalassemia? Blood 94. no. 9: 3199–3204.
Rosenbloom, K. R., C. A. Sloan, V. S. Malladi, T. R. Dreszer, K. Learned, V. M. Kirkup, M. C. Wong, et al. 2013. ENCODE data in the UCSC genome browser: Year 5 update. Nucleic Acids Research 41. doi: 10.1093/nar/gks1172.
Roy, P., G. Bhattacharya, A. Mandal, U. B. Dasqupta, D. Banerjee, S. Chandra, and M. Das. 2012. Influence of BCL11A, HBS1L-MYB, HBBP1 single nucleotide polymorphisms and the HBG2 XmnI polymorphism on Hb F levels. Hemoglobin 36, no. 6: 592–599.
Sankaran, V. G., J. Xu, and S. H. Orkin. 2010. Advances in the understanding of haemoglobin switching. British Journal of Haematology 149, no. 2: 181–194.
Sheffield, N. C., R. E. Thurman, L. Song, A. Safi, J. A. Stamatoyannopoulos, B. Lenhard, G. E. Crawford, and T. S. Furey. 2013. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Research 23, no. 5: 777–788.
Svensson, Ö., L. Arvestad, and J. Lagergren. 2006. Genome-wide survey for biologically functional pseudogenes. PLoS Computational Biology 2, no. 5: e46.
Thurman R. E., E. Rynes, R. Humbert, J. Vierstra, M. T. Maurano, E. Haugen, N. C. Sheffield, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489, no. 7414: 75–82.
Tutar, Y. 2012. Pseudogenes. Comparative and Functional Genomics. doi: 10.1155/2012/424526.
Wang, Z., C. Zang, J. A. Rosenfeld, D. E. Schones, A. Barski, S. Cuddapah, K. Cui, et al. 2008. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature Genetics 40: 897–903.
Wen. Y. Z., L. L. Zheng, L. H. Qu, F. J. Ayala, and Z. R. Lun. 2012. Pseudogenes are not pseudo any more. RNA Biology 9, no. 1: 27–32.
Xu, J., V. G. Sankaran, M. Ni, T. F. Menne, R. V. Puram, W. Kim and S. H. Orkin. 2010. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes and Development 24, no. 8: 783–798.
Xu, J., D. E. Bauer, M. A. Kerenyi, T. D. Vo, S. Hou, Y.-J. Hsu, H. Yao, J. J. Trowbridge, G. Mandel, and S. H. Orkin. 2012. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proceedings of the National Academy of Sciences of the United States of America 110, no. 16: 6518–6523.
Zentner, G. E., P. J. Tesar, and P. C. Scacheri. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Research 21, no. 8: 1273–1283.