The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
Evasion of cytotoxic T lymphocytes is a major driving force of HIV-1 evolution within a host. In a genetically homogenous population where some MHC class I types are dominant, repeated selection of escape mutants can cause HIV-1 to lose fitness. However, in a heterogenous population, reversions are more frequent and attenuation of HIV is slower. HIV-1 may have experienced adaptation to the human host after crossing species from primates, but immune selection and random drift are causing the viral genome to degenerate. Antiviral therapy further cuts into viral fitness, even when the drugs are resisted. HIV-1 is an important example which shows that genetic entropy is operating throughout the biological realm, even while meaningful genetic adaptations are occurring.
Keywords: HIV-1, fitness, genetic entropy
Introduction
Human immunodeficiency virus type 1 (HIV-1), causative agent of the AIDS pandemic, is notorious for rapidly accumulating mutations due to its error-prone RNA polymerase and the absence of RNA repair enzymes. However, long-term maintenance of proviral DNA in resting host cells prevents the viral genome from rapid degeneration (Salgado et al. 2010). The diploid nature of the viral genome and frequent recombination also helps to preserve lentiviral lineages. The situation is analogous to the stability of diploid germline genomes in higher organisms because reproductive cells experience fewer cycles of cell division and undergo regular genetic recombination during meiosis. Nevertheless, we know that even higher genomes are degenerating (Sanford 2014).
Since its introduction into mankind in the beginning of the twentieth century (Sharp and Hahn 2011; Wertheim and Worobey 2009), HIV-1 has replicated in humans through a great many generations—which is comparable with deep evolutionary time in higher organisms (Behe 2007). Therefore, analyzing the evolution of HIV-1 through the past few decades offers a glimpse into the evolution of diploid cellular genomes through evolutionary time. The major forces that shape the HIV-1 genome are immune evasion, random drift, and antiretroviral therapy.
Racing to Stay Ahead of Host Antibodies
The impressive reproduction rate of HIV-1 (up to 1010 viral particles daily, Goering et al. 2013) affords strong selection. The most important selective force in untreated patients is host immunity. The infected host produces antibodies, especially antibodies against the surface glycoprotein, Env, which can neutralize extracellular viral particles (Wei et al. 2003). However, mutations in the env gene of HIV-1 quickly render the antibodies powerless. The host can produce new antibodies against the changing virus, but viral evolution is always faster than antibody development. Even though natural selection generally results in directional evolution, the random nature of antibody development and the recurrent nature of HIV mutation results in cyclic selection (Shriner et al. 2004). In the long run, antibodies do not drive the virus anywhere specific. Conceivably, some mutants have compromised replication capacity (fitness), but when the virus is transmitted into another host, antibody-driven mutations will likely revert as higher replication capacity is favored.
War of Attrition Against T lymphocytes
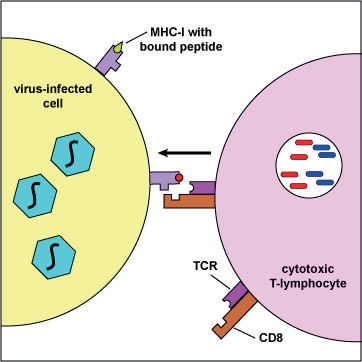
Fig. 1. Presentation of viral peptides by an infected cell to a cytotoxic T lymphocyte, using the MHC-I complex. (Illustration by Kaiser 2008).
In HIV infection, antibodies are not nearly as protective as cytotoxic T lymphocytes (CTLs), which attack and kill HIV-infected cells. In contrast to the almost unlimited number of antibodies that a body can produce, the CTL response is limited by selective presentation of viral peptides (epitopes) on the surface of the infected cell, using MHC class I molecules as carriers (see fig. 1 after Kaiser 2008). Each person has a unique set of six types of MHC-I molecules. The uniqueness is due to diverse forms of MHC genes in the population. The MHC-I molecules of an infected individual presents a unique repertoire of HIV peptides to his/her CTL cells. However, the overall number of HIV peptides presentable by human MHC-I molecules is limited (a few hundred, see the official list in table 1, reviewed by Llano et al. 2013).
| Epitope | Protein | HXB2 Location | Subprotein | HXB2 DNA Contig | Subtype | Species | HLA |
|---|---|---|---|---|---|---|---|
| GELDRWEKI | Gag | 11-19 | s>p17(11-19) | 820..846 | human | B*4002 | |
| KIRLRPGGK | Gag | 18-26 | s>p17(18-26) | 841..867 | human | A*0301 | |
| IRLRPGGKK | Gag | 19-27 | s>p17(19-27) | 844..870 | B | human | B*2705 |
| RLRPGGKKK | Gag | 20-28 | p17(20-28) | 847..873 | human | A*0301 | |
| RLRPGGKKKY | Gag | 20-29 | s>p17(20-29) | 847..876 | B | human | A*0301 |
| RPGGKKKYKL | Gag | 22-31 | s>p17(22-31) | 853..882 | B | human | B*5101 |
| GGKKKYKLK | Gag | 24-32 | s>p17(24-32) | 859..885 | B | human | B*0801 |
| KYKLKHIVW | Gag | 28-36 | s>p17(28-36) | 871..897 | B | human | A*2402 |
| HLVWASREL | Gag | 33-41 | s>p17(33-41) | 886..912 | human | Cw*0804 | |
| LVWASRELERF | Gag | 34-44 | s>p17(34-44) | 889..921 | B | human | A30 |
| WASRELERF | Gag | 36-44 | s>p17(36-44) | 895..921 | B | human | B*3501 |
| ELRSLYNTV | Gag | 74-82 | s>P17(74-82) | 1009..1035 | human | B*0801 | |
| RSLYNTVATLY | Gag | 76-86 | s>p17(76-86) | 1015..1047 | B | human | A*3002, B58, B63 |
| SLYNTVATL | Gag | 77-85 | s>p17(77-85) | 1018..1044 | B | human | A*0201, A*0202, A*0205 |
| SLYNTVATLY | Gag | 77-86 | s>p17(77-86) | 1018..1047 | B | human | A*0201 |
| LYNTVATL | Gag | 78-85 | s>p17(78-85) | 1021..1044 | human | Cw14 | |
| LYNTVATLY | Gag | 78-86 | s>p17(78-86) | 1021..1047 | human | A*2092, B*4403 | |
| TLYCVHOK | Gag | 84-91 | s>p17(84-91) | 1039..1062 | human | A*1101 | |
| IEIKDTKEAL | Gag | 92-101 | s>p17(92-101) | 1063..1092 | human | B*4001 | |
| NSSKVSONY | Gag | 124-132 | s>p17(124-132) | 1159..1185 | B | human | B*3501 |
| VONLOGOMV | Gag | 135-143 | s>p24(3-11) | 1192..1218 | human | B13 | |
| HOAISPRTL | Gag | 144-152 | s>p24(12-20) | 1219..1245 | human | B*1510 | |
| OAISPRTLNAW | Gag | 145-155 | s>p24(13-23) | 1222..1254 | B | human | A*2501 |
| ISPRTLNAW | Gag | 147-155 | s>p24(15-23) | 1228..1254 | human | B*5701, B63 | |
| SPRTLNAWV | Gag | 148-156 | s>p24(16-24) | 1231..1257 | human | B*0702 | |
| VKVIEEKAF | Gag | 156-164 | s>p24(24-32) | 1255..1281 | human | B*1503 | |
| EEKAFSPEV | Gag | 160-168 | s>p24(28-36) | 1267..1293 | human | B*4415 | |
| KAFSPEVI | Gag | 162-169 | s>p24(30-37) | 1273..1296 | B | human | B*5703 |
| KAFSPEVIPMF | Gag | 162-172 | s>p24(30-40) | 1273..1305 | B | human | B*5701, B*5703, B63 |
| FSPEVIPMF | Gag | 164-172 | s>p24(32-40) | 1279..1305 | human | B57 | |
| EVIPMFSAL | Gag | 167-175 | s>p24(35-43) | 1288..1314 | B | human | A*2601, A*2602, A*2603 |
| VIPMFSAL | Gag | 168-175 | s>p24(36-43) | 1291..1314 | B | human | Cw*0102 |
| SEGATPODL | Gag | 176-184 | s>p24(44-52) | 1315..1341 | human | B*4001 | |
| TPODLNTML | Gag | 180-188 | s>p24(48-56) | 1327..1353 | B | human | B*0702, B*3910, B*4201, B*8101, Cw*0802 |
| TPODLNMML | Gag | 180-188 | s>p24(48-56) | 1327..1353 | A | human | B53 |
| TPYDINOML | Gag | 180-188 | s>p24(48-56) | 1327..1353 | HIV-2 | human | B*5301 |
| GHOAAMOML | Gag | 193-201 | s>p24(61-69) | 1366..1392 | B | human | B*1510, B*3901 |
| KETINEEAA | Gag | 202-210 | p24(70-78) | 1393..1419 | human | B*4002 | |
| ETINEEAAEW | Gag | 203-212 | p24(71-80) | 1396..1425 | human | A*2501 | |
| AEWDRVHPV | Gag | 210-218 | p24(78-86) | 1417..1443 | human | B*4002 | |
| HPVHAGPIA | Gag | 216-224 | p24(84-92) | 1435..1461 | human | B*3501, B7 | |
| GOMREPRGSDI | Gag | 226-236 | p24(94-104) | 1465..1497 | human | B13 | |
| TSTLOEOIGW | Gag | 240-249 | p24(108-117) | 1507..1536 | B | human | B*5701, B*5801 |
| NPPIPVGDIY | Gag | 253-262 | p24(121-130) | 1546..1575 | human | B*3501 | |
| PPIPVGDIY | Gag | 254-262 | p24(122-130) | 1549..1575 | B | human | B*3051 |
| EIYKRWII | Gag | 260-267 | p24(128-135) | 1567..1590 | B | human | B*0801 |
| RRWIOLGLOK | Gag | 263-272 | p24(131-140) | 1576..1605 | human | B*2703 | |
| KRWIILGLNK | Gag | 263-272 | p24(131-140) | 1576..1605 | B | human | B*2705 |
| GLNKIVRMY | Gag | 269-277 | p24(137-145) | 1594..1620 | B | human | B*1501, B62 |
| VRMYSPVSI | Gag | 274-282 | p24(142-150) | 1609..1635 | human | Cw18 | |
| RMYSPTSI | Gag | 275-282 | p24(143-150) | 1612..1635 | human | B*5201 | |
| YSPVSILDI | Gag | 277-285 | p24(145-153) | 1618..1644 | CRF01_AE | human | Cw*0102 |
| FRDYVDRFF | Gag | 293-301 | p24(161-169) | 1666..1692 | human | Cw18 | |
| FRDYVDRFYK | Gag | 293-302 | p24(161-170) | 1666..1695 | B, D | human | B*1801 |
| RDYVDRFFKTL | Gag | 294-304 | p24(162-172) | 1669..1701 | A | human | A*2402 |
| RDYVDRFYKTL | Gag | 294-304 | p24(162-172) | 1669..1701 | B | human | B*4402 |
| YVDRFYKTL | Gag | 296-304 | p24(164-172) | 1675..1701 | human | A*0207 | |
| YVDRFFKTL | Gag | 296-304 | p24(164-172) | 1675..1701 | human | B*1503, Cw*0303, Cw*0304 |
|
| DRFYKTLRA | Gag | 298-306 | p24(166-174) | 1681..1707 | B | human | B*1402 |
| AEOASODVKNW | Gag | 306-316 | p24(174-184) | 1705..1737 | B | human | B*4402 |
| AEOASOEVKNWM | Gag | 306-317 | p24(174-185) | 1705..1740 | human | Cw5 | |
| OASOEVKNW | Gag | 308-316 | p24(176-184) | 1711..1737 | B | human | B*5301, B*5701, B*5801 |
| VKNWMTETL | Gag | 313-321 | p24(181-189) | 1726..1752 | B | human | B*4801 |
| DCKTILKAL | Gag | 329-337 | p24(197-205) | 1774..1800 | B | human | B*0801 |
| ACOGVGGPGHK | Gag | 349-359 | p24(217-227) | 1834..1866 | human | A*1101 | |
| GPGHKARVL | Gag | 355-363 | p24(223-231) | 1852..1878 | B | human | B*0702 |
| AEAMSOVTNS | Gag | 364-373 | p2p7p1p6(1-10) | 1879..1908 | human | B*4501 | |
| CRAPRKKGC | Gag | 405-413 | p2p7p1p6(42-50) | 2002..2028 | human | B14 | |
| TEROANFL | Gag | 427-434 | p2p7p1p6(64-71) | 2068..2091 | human | B*4002 | |
| ROANFLGKI | Gag | 429-437 | p2p7p1p6(66-74) | 2074..2100 | B | human | B*4801, B13 |
| FLGKIWPSYK | Gag | 433-442 | p2p7p1p6(70-79) | 2086..2115 | human | A*0201 | |
| KELYPLTSL | Gag | 481-489 | p2p7p1p6(118-126) | 2230..2256 | human | B*4001 | |
| NSPTRREL | Pol | 24-31 | Gag/Pol-TF(24-31) | 2154..2177 | human | Cw*0102 | |
| ITLWORPLV | Pol | 59-67 | Protease(3-11) | 2259..2285 | A, B, D | human | A*6802, A*7401 |
| DTVLEEWNL | Pol | 86-94 | Protease(30-38) | 2340..2366 | C | human | A*6802 |
| EEMNLPGRW | Pol | 90-98 | Protease(34-42) | 2352..2378 | human | B44 | |
| ROYDOILIEI | Pol | 113-122 | Protease(57-66) | 2421..2450 | human | B13 | |
| GKKAIGTVL | Pol | 124-132 | Protease(68-76) | 2454..2480 | human | B*1503 | |
| KAIGTVLV | Pol | 126-133 | Protease(70-77) | 2460..2483 | human | B57 | |
| LVGPTPVNI | Pol | 132-140 | Protease(76-84) | 2478..2504 | human | A*0201 | |
| TPVNIIGRNML | Pol | 136-146 | Protease(80-90) | 2490..2522 | human | B81 | |
| FPISPIETV | Pol | 155-163 | Protease(99)-RT(8) | 2547..2573 | B | human | B*5401 |
| IETVPVKL | Pol | 160-167 | RT(5-12) | 2562..2585 | human | B*4001 | |
| GPKVKOWPL | Pol | 173-181 | RT(18-26) | 2601..2627 | B | human | B*0801 |
| ALVEICTEM | Pol | 188-196 | RT(33-41) | 2646..2672 | B | human | A*0201 |
| ALVEICTEMEK | Pol | 188-198 | RT(33-43) | 2646..2678 | human | A*0301 | |
| KLVDFRELNK | Pol | 228-237 | RT(73-82) | 2766..2795 | human | A*0301 | |
| GIPHPAGLK | Pol | 248-256 | RT(93-101) | 2826..2852 | B | human | A*0301 |
| TVLDVGDAY | Pol | 262-270 | RT(107-115) | 2868..2894 | human | B*3501 | |
| VPLDEDFRKY | Pol | 273-282 | RT(118-127) | 2901..2930 | human | B*3501 | |
| YTAFTIPSV | Pol | 282-290 | RT(127-135) | 2928..2954 | human | A2 | |
| TAFTIPSI | Pol | 283-290 | RT(129-135) | 2931..2954 | human | B*5101 | |
| NETPGIRYOY | Pol | 292-301 | RT(137-146) | 2958..2987 | human | B18 | |
| TRYOYNVL | Pol | 297-304 | RT(142-149) | 2973..2996 | human | B*1401 | |
| LPOGWKGSPA | Pol | 304-313 | RT(149-158) | 2994..3023 | B | human | B*5401 |
| SPAIFOSSM | Pol | 311-319 | RT(156-164) | 3015..3041 | human | B7 | |
| AIFOSSMTK | Pol | 313-321 | RT(158-166) | 3021..3047 | B | human | A*0301, A*1101 |
| KONPDIVIY | Pol | 328-336 | RT(173-181) | 3066..3092 | B | human | A*3002, Cw*1202 |
| NPEIVIYOY | Pol | 330-338 | RT(175-183) | 3072..3098 | human | B18 | |
| HPDIVIYOY | Pol | 330-338 | RT(175-183) | 3072..3098 | B | human | B*3501 |
| VIYOYMDDL | Pol | 334-342 | RT(179-187) | 3084..3110 | B | human | A*0201 |
| IEELROHLL | Pol | 357-365 | RT(202-210) | 3153..3179 | B | human | B*4001 |
| IVLPEKDSW | Pol | 399-407 | RT(244-252) | 3279..3305 | human | B*5701 | |
| LVGKLNWASOIY | Pol | 415-426 | RT(260-271) | 3327..3362 | human | B*1501 | |
| KLNWASOIY | Pol | 418-426 | RT(263-271) | 3336..3362 | B | human | A*3002 |
| OIYPGIKVR | Pol | 424-432 | RT(269-277) | 3354..3380 | B | human | A*0301 |
| YPGIKVROL | Pol | 426-434 | RT(271-279) | 3360..3386 | B | human | B*4201 |
| IPLTEEAEL | Pol | 448-456 | RT(293-301) | 3426..3452 | human | B*3501, B*5101 |
|
| ILKEPVHGV | Pol | 464-472 | RT(309-317) | 3474..3500 | B | human | A*0201 |
| ILKEPVHGVY | Pol | 464-473 | RT(309-318) | 3474..3503 | B | human | B*1501, Cw*1202 |
| GOGOWTYOI | Pol | 488-496 | RT(333-341) | 3546..3572 | human | B13 | |
| IYOEPFKNLK | Pol | 496-505 | RT(341-350) | 3570..3599 | B | human | A*1101 |
| RMRGAHTNDV | Pol | 511-520 | RT(356-365) | 3615..3644 | human | A*3002 | |
| RMRGAHTNDVK | Pol | 511-521 | RT(356-366) | 3615..3647 | human | A*0301 | |
| IAMESIVIW | Pol | 530-538 | RT(375-383) | 3672..3698 | human | B*5801 | |
| PIOKETWETW | Pol | 547-556 | RT(392-401) | 3723..3752 | B | human | A*3201 |
| GAETFYVDGA | Pol | 591-600 | RT(436-445) | 3855..3884 | human | A*6802 | |
| ETFYVDGAANR | Pol | 593-603 | RT(438-448) | 3861..3893 | human | A66 | |
| ETKLGKAGY | Pol | 604-612 | RT(449-457) | 3894..3920 | human | A*2601 | |
| IVTDSOYAL | Pol | 650-658 | RT(495-503) | 4032..4058 | human | Cw*0802 | |
| VTDSOYALGI | Pol | 651-660 | RT(496-505) | 4035..4064 | human | B*1503 | |
| OIIEOLIKK | Pol | 675-683 | RT(520-528) | 4107..4133 | B | human | A*1101 |
| LFLDGIDKA | Pol | 715-723 | RT(560)-Integrase(8) | 4227..4253 | human | B81 | |
| LPPIVAKEI | Pol | 743-751 | Integrase(28-36) | 4311..4337 | human | B*4201 | |
| THLEGKIIL | Pol | 781-789 | Integrase(66-74) | 4425..4451 | human | B*1510 | |
| HVASGYIEA | Pol | 793-801 | Integrase(78-86) | 4461..4487 | B | human | B*5401 |
| IEAEVIPAET | Pol | 799-808 | Integrase(84-93) | 4479..4508 | B | human | B*4002 |
| HTDNGSNF | Pol | 829-836 | Integraser(114-121) | 4569..4592 | human | Cw5 | |
| STTVKAACWW | Pol | 838-847 | Integrase(123-132) | 4596..4625 | human | B57 | |
| IOOEFGIPY | Pol | 850-858 | Integrase(135-143) | 4632..4658 | human | B*1503 | |
| VRDOAEHL | Pol | 880-887 | Integrase(165-172) | 4722..4745 | human | Cw18 | |
| KTAVOMAVF | Pol | 888-896 | Integrase(173-181) | 4746..4772 | human | B*5701 | |
| AVFIHNFKRK | Pol | 894-903 | Integrase(179-188) | 4764..4793 | B | human | A*0301, A*1101 |
| FKRKGGIGGY | Pol | 900-909 | Integrase(185-194) | 4782..4811 | human | B*1503 | |
| KRKGGIGGY | Pol | 901-909 | Integrase(186-194) | 4785..4811 | human | B*2705 | |
| GERIVDII | Pol | 912-919 | Integrase(197-204) | 4818..4841 | B | human | B*4002 |
| IIATDIOTK | Pol | 918-926 | Integrase(203-211) | 4836..4862 | B | human | A11 |
| KIONFRVYY | Pol | 934-942 | Integrase(219-277) | 4884..4910 | human | A*3002 | |
| VPRRKAKII | Pol | 975-983 | Integrase(260-268) | 5007..5033 | human | B42 | |
| RKAKIIRDY | Pol | 978-986 | Integrase(263-271) | 5016..5042 | human | B*1503 | |
| RIRTWKSLVK | Vif | 17-26 | 5089..5118 | B | human | A*0301 | |
| HMYISKKAK | Vif | 28-36 | 5122..5148 | human | A*0301 | ||
| ISKKAKGWE | Vif | 31-39 | 5131..5157 | human | B*5701 | ||
| HPRVSSEVHI | Vif | 48-57 | 5182..5211 | human | B*0702 | ||
| IPLGDAKLII | Vif | 57-66 | 5209..5238 | human | B51 | ||
| WHLGHGVSI | Vif | 79-87 | 5275..5301 | human | B*1510 | ||
| WHLGOGVSI | Vif | 79-87 | 5275..5301 | human | B*3801 | ||
| LGHGVSIEW | Vif | 81-89 | 5281..5307 | human | B*5703 | ||
| LADOLIHLHY | Vif | 102-111 | 5344..5373 | human | B*1801 | ||
| KTKPPLPSVKK | Vif | 158-168 | 5512..5544 | human | A*0301 | ||
| EAVRHFPRI | Vpr | 29-37 | 5643..5669 | human | B51 | ||
| AVRHFPRIW | Vpr | 30-38 | 5646..5672 | human | B*5701 | ||
| VRHFPRIWL | Vpr | 31-39 | 5649..5675 | human | B27 | ||
| FPRIWLHGL | Vpr | 34-42 | 5658..5684 | human | B*0702, B*8101 |
||
| ETYGDTWTGV | Vpr | 48-57 | 5700..5729 | human | A*6802 | ||
| DTWAGVEAIIR | Vpr | 52-62 | 5712..5744 | human | A*6801 | ||
| AIIRILOOL | Vpr | 59-67 | 5733..5759 | B | human | A*0201 | |
| CCFHCOVC | Tat | 30-37 | 5918..5941 | human | Cw12 | ||
| FOTKGLGISY | Tat | 38-47 | 5942..5971 | human | B*1503 | ||
| ITKGLGISYGR | Tat | 39-49 | 5945..5977 | human | A*6801 | ||
| KAVRLIKFLY | Rev | 14-23 | 6009..6038 | B | human | B*5701, B*5801, B63 |
|
| OAVRIIKILY | Rev | 14-23 | 6009..6038 | C | human | B*5703 | |
| ERILSTYLGR | Rev | 57-66 | 8471..8500 | human | A*0301 | ||
| RPAEPVPLOL | Rev | 66-75 | 8498..8527 | human | B7 | ||
| SAEPVPLOL | Rev | 67-75 | 8501..8527 | B | human | Cw*0501 | |
| YRLGVGALI | Vpu | 5-13 | 6074..6100 | C | human | Cw18 | |
| EYRKILROR | Vpu | 29-37 | 6146..6172 | human | A*3303 | ||
| RVKEKYOHL | gp160 | 2-10 | gp120(2-10) | ||||
| AENLWVTVY | gp160 | 31-39 | gp120(31-39) | 6315..6341 | human | B*1801, B44 |
|
| AENLWVTVYY | gp160 | 31-40 | gp120(31-40) | 6315..6344 | human | B*4402 | |
| TVYYGVPVWK | gp160 | 37-46 | gp120(37-46) | 6333..6362 | B | human | A*0301 |
| VPVWKEATTT | gp160 | 42-51 | gp120(42-51) | 6348..6377 | human | B*5501 | |
| VPVWKEATTTL | gp160 | 42-52 | gp120(42-52) | 6328..6380 | human | B*3501 | |
| LFCASDAKAY | gp160 | 52-61 | gp120(52-61) | 6378..6407 | B | human | A*2402 |
| KAYETEVHNVW | gp160 | 59-69 | gp120(59-69) | 5399..6431 | human | B58 | |
| YETEVHNVW | gp160 | 78-86 | gp120(78-86) | 6456..6482 | human | B*3501 | |
| MHEDIISLW | gp160 | 104-112 | gp120(104-112) | 6534..6560 | human | B*3801 | |
| SVITOACPK | gp160 | 199-207 | gp120(199-207) | 6819..6845 | human | A*1101 | |
| SFEPIPIHY | gp160 | 209-217 | gp120(209-217) | 6849..6875 | B | human | A*2902 |
| CAPAGFAIL | gp160 | 218-226 | gp120(218-226) | 6876..6902 | human | Cw1 | |
| RPNNNTRKSI | gp160 | 298-307 | gp120(298-307) | 7116..7145 | B | human | B*0702 |
| HIGPGRAFY | gp160 | 310-318 | gp120(310-318) | 7152..7178 | human | A*3002 | |
| RGPGRAFVTI | gp160 | 311-320 | gp120(311-320) | 7155..7184 | human | A*0201 | |
| EIIGDIROAY | gp160 | 321-330 | gp120(321-330) | 7185..7214 | human | A*2501 | |
| SFNCGGEFF | gp160 | 375-383 | gp120(375-383) | 7374..7373 | B | human | B*1516, Cw*0401 |
| LPCRIKOII | gp160 | 416-424 | gp120(416-424) | 7470..7496 | B | human | B*5101 |
| RIKOIINMW | gp160 | 419-427 | gp120(419-427) | 7479..7505 | B | human | A*3201 |
| RAIEAOOHL | gp160 | 557-565 | gp41(46-54) | 7893..7919 | human | Cw*0304, Cw15 |
|
| RAIEAOOHM | gp160 | 557-565 | gp41(46-54) | 7893..7919 | human | Cw8 | |
| OTRVLAIERYL | gp160 | 577-587 | gp41(66-76) | 7953..7985 | C | human | B*5802 |
| ERYLKDOOL | gp160 | 584-592 | gp41(73-81) | 7974..8000 | human | B*1402 | |
| RYLKDOOLL | gp160 | 585-593 | gp41(74-82) | 7977..8003 | B | human | A*2402, A23 |
| YLKDOOLL | gp160 | 586-593 | gp41(75-82) | 7980..8003 | human | B*0801 | |
| TAVPWNASW | gp160 | 606-614 | gp41(95-103) | 8040..8066 | B | human | B*3501 |
| VFAVLSIVNR | gp160 | 698-707 | gp41(187-196) | 8316..8345 | human | A*3303 | |
| IVNRNROGY | gp160 | 704-712 | gp41(193-201) | 8334..8360 | B | human | A*3002 |
| RLRDLLLIVTR | gp160 | 770-780 | gp41(259-269) | 8532..8564 | B | human | A*0301, A*3101 |
| IVTRIVELL | gp160 | 777-785 | gp41(266-274) | 8553..8579 | B | human | A*6802 |
| GRRGWEALKY | gp160 | 786-795 | gp41(275-284) | 8580..8609 | B | human | B*2705 |
| RRGWEVLKY | gp160 | 787-795 | gp41(276-284) | 8583..8609 | human | A*0101 | |
| KYCWNLLOY | gp160 | 794-802 | gp41(283-291) | 8604..8630 | B | human | A*3002 |
| OELKNSAVSL | gp160 | 805-814 | gp41(294-303) | 8637..8666 | B | human | B*4001 |
| SLLNATDIAV | gp160 | 813-822 | gp41(302-311) | 8661..8690 | B | human | A*0201 |
| LLNATDIAV | gp160 | 814-22 | gp41(303-311) | 8664..8690 | B | human | A*0201 |
| EVAORAYR | gp160 | 831-838 | gp41(320-327) | 8715..8738 | human | A*3303 | |
| IPRRIROGL | gp160 | 843-851 | gp41(332-340) | 8751..8777 | B | human | B*0702 |
| RIROGLERA | gp160 | 846-854 | gp41(335-343) | 8760..8786 | human | A*0205 | |
| ROGLERALL | gp160 | 848-856 | gp41(337-345) | ||||
| WPTVRERM | Nef | 13-20 | 8833..8856 | B | human | B*0801 | |
| RMRRAEPAA | Nef | 19-27 | 8851..8877 | human | B62 | ||
| LEKHGAITS | Nef | 37-45 | 8905..8931 | human | B*4001, B50 |
||
| FPVTPOVPL | Nef | 68-76 | 8998..9024 | human | B*0702 | ||
| FPVTPOVPLR | Nef | 68-77 | 8998..9027 | B | human | B+0702 | |
| TPOVPLRPM | Nef | 71-79 | 9007..9033 | B | human | B*0702 | |
| RPOVPLRPM | Nef | 71-79 | 9007..9033 | human | B*4201, B*4202 |
||
| RPOVPLRPMTY | Nef | 71-81 | 9007..9039 | B | human | B35 | |
| OVPLRPMTYK | Nef | 73-82 | 9013..9042 | B | human | A*0301, A*1101 |
|
| VPLRPMTY | Nef | 74-81 | 9016..9039 | B | human | B*3501 | |
| PLRPMTYK | Nef | 75-82 | 9019..9042 | B | human | A*1101 | |
| LRPMTYKAA | Nef | 76-84 | 9022..9048 | B | human | B*2703 | |
| RPMTYKAAL | Nef | 77-85 | 9025..9051 | B | human | B*0702 | |
| KAAFDLSFF | Nef | 82-90 | 9040..9066 | human | B*5703, B*5801 |
||
| KAAVDLSHFL | Nef | 82-91 | 9040..9069 | human | Cw8 | ||
| GAFDLSFFL | Nef | 83-91 | 9043..9069 | human | A*0205 | ||
| AAFDLSFFL | Nef | 83-91 | 9043..9069 | human | B*5703 | ||
| AAVDLSHFL | Nef | 83-91 | 9043..9069 | B | human | Cw*0802 | |
| AALDLSHFL | Nef | 83-91 | 9043..9069 | human | Cw3 | ||
| AVDLSHFLK | Nef | 84-92 | 9046..9072 | B | human | A*0301, A*1101 |
|
| FLKEKGGL | Nef | 90-97 | 9064..9087 | B | human | B*0801 | |
| KEKGGLEGL | Nef | 92-100 | 9070..9096 | B | human | B*4001, B*4002 |
|
| RRODILDLWI | Nef | 105-114 | 9109..9138 | B | human | B*2705 | |
| RRODILDLWVY | Nef | 105-115 | 9109..9141 | human | B18 | ||
| KROEILDLWVY | Nef | 105-115 | 9109..9141 | human | Cw7 | ||
| RODILDLWI | Nef | 106-114 | 9112..9138 | human | B13 | ||
| RODILDLWV | Nef | 106-114 | 9112..9138 | human | B*1302 | ||
| HTOGYFPDW | Nef | 116-124 | 9142..9168 | human | B*5703, B*5801, B57 |
||
| TOGYFPDWONY | Nef | 117-127 | 9145..9177 | B | human | B*1501 | |
| YFPDWONYT | Nef | 120-128 | 9154..9180 | B | human | A29, B*3501, Cw6 |
|
| FPDWONYTP | Nef | 121-129 | 9157..9183 | B | human | B*5401 | |
| YTPGPGIRY | Nef | 127-135 | 9175..9201 | human | B57, B63 | ||
| TPGPGVRYPL | Nef | 128-137 | 9178..9207 | B | human | B*0702, B*4201, B*4202 |
|
| TRYPLTFGW | Nef | 133-141 | 9193..9219 | human | A33 | ||
| RYPLTFGW | Nef | 134-141 | 9196..9219 | B | human | A*2402 | |
| YPLTFGWCY | Nef | 135-143 | 9199..9225 | B | human | B*1801, B*5301 |
|
| YPLTFGWCF | Nef | 135-143 | 9199..9225 | human | B53 | ||
| PLTFGWCYKL | Nef | 136-145 | 9202..9231 | B | human | A*0201 | |
| LTFGWCFKL | Nef | 137-145 | 9205..9231 | human | B57, B63 | ||
| VLEWRFDSRL | Nef | 180-189 | 9334..9363 | B | human | A*0201 | |
| WRFDSRLAF | Nef | 183-191 | 9343..9369 | human | B*1503 |
During the early stages of infection, the CTL response successfully brings HIV replication under control, but some viruses always escape by mutating CTL targets. If the HIV peptides presented by the MHC-I molecules happen to be located on viral protein domains critical for replication, escape mutants will suffer lowered replication capacity (fitness loss). In such patients, there will be a long war of attrition between the immune system and the virus (Boutwell, Rowley, and Essex 2009). On the other hand, if the HIV peptides presented by the MHC-I molecules are few in number and/or located on less critical regions of HIV proteins, the virus will escape the CTLs easily without significant fitness cost. The virus will destroy infected T lymphocytes, overpower the CTLs, and cause AIDS after a short latency period (Yue et al. 2015).
Most patients are infected by a single viral particle or a single infected cell (Keele et al. 2008). The virus rapidly diversifies within the host. Subsequent immune selection curbs the rate of viral diversification (see fig. 2, after Maldarelli et al. 2013). Contrary to Darwinian thinking, the replication capacity of the early viral population is high, and quickly declines along an exponential curve (see fig. 3, after Arnott et al. 2010), presumably due to accumulation of escape mutants with compromised fitness. However, as recognizable CTL targets dwindle, and as CD4+ T cells are killed by the virus, the virus gradually diversifies again, with concurrent increase in replication capacity and plasma viral load (Arnott et al. 2010; Maldarelli et al. 2013; Troyer et al. 2005). The relationship between viral fitness, diversity, and plasma viral load is schematically illustrated in Fig. 4.
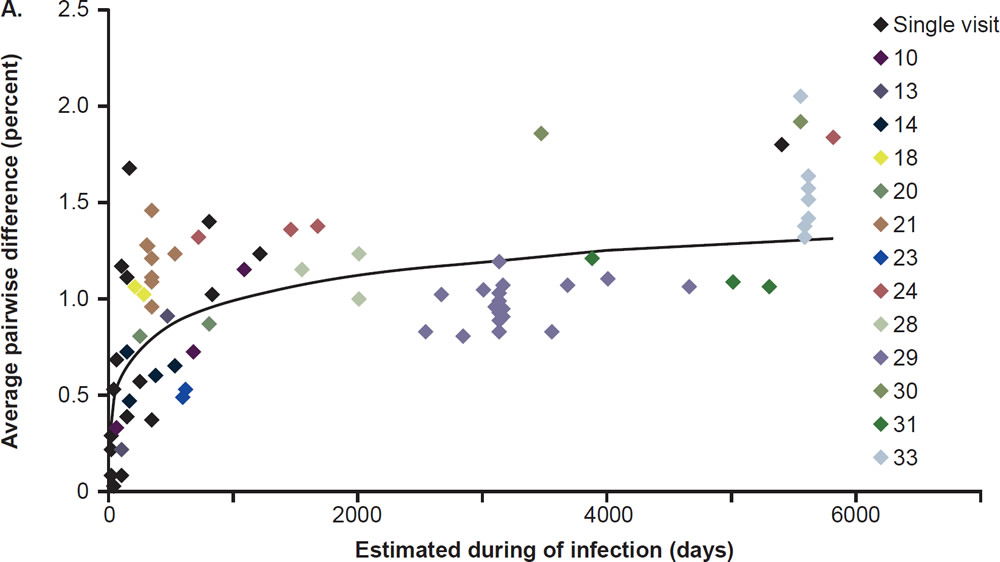
Fig. 2. Nonlinear accumulation of HIV diversity over time. Overall diversity was determined from alignments of pro-pol sequences. Multiple samples from the same patient are shown with the same color. Patients for whom only a single sample was available for analysis are shown in black. After Fig. 2A of Maldarelli et al. 2013 at http://www. ncbi.nlm.nih.gov/pmc/articles/PMC3754011/figure/F2/.
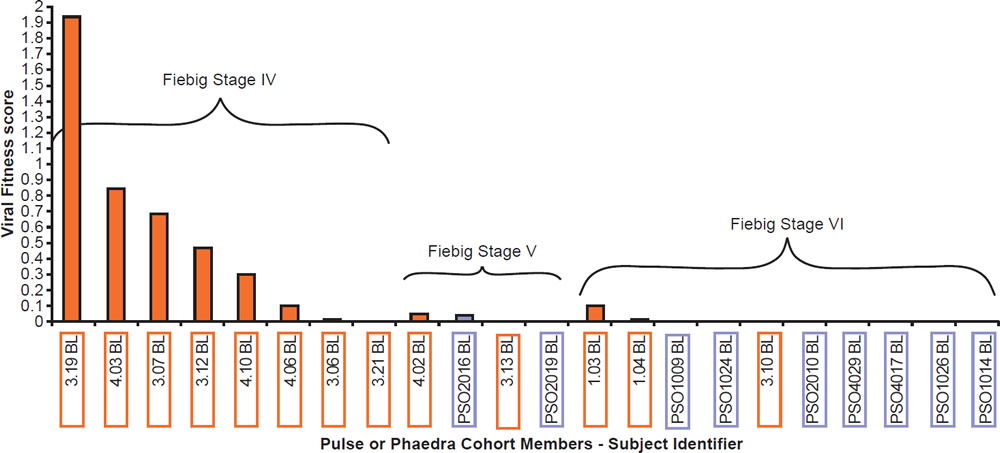
Fig. 3. Viral fitness of baseline isolates obtained from acute HIV-1 infection subjects (PULSE) and from early chronic HIV-1 infection subjects (PHAEDRA) relative to the stage of seroconversion, after Fig. 10 of Arnott et al. 2010 at http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2936565/figure/pone-0012631-g010/.
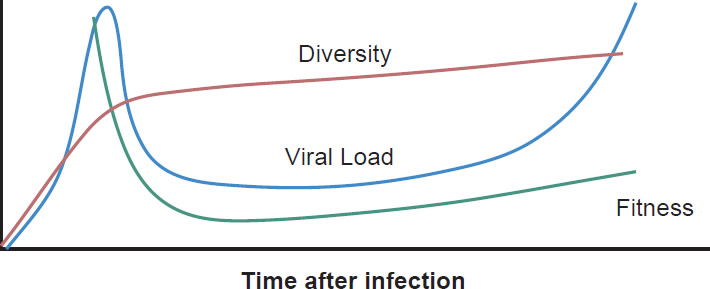
Fig. 4. Viral diversity, fitness, and plasma viral load during HIV-1 infection. The diversity curve is based on Fig. 2A of Maldarelli et al. 2013. The fitness curve is based on Fig. 10 of Arnott et al. 2010. The viral load curve is based on Fig. 4 of an article on the website of the National Institute of Allergy and Infectious Diseases entitled “The Relationship Between the Human Immunodeficiency Virus and the Acquired Immunodeficiency Syndrome”. http://www.niaid.nih.gov/topics/hivaids/understanding/howhivcausesaids/pages/relationshiphivaids.aspx.
Long-Term Evolution
If a population is genetically homogenous, individuals will have similar MHC phenotypes and their CTLs would target similar HIV peptides. Passage of the virus through the population will consistently select for the same immune escape mutations. Although the resulting mutants are adapted to protective CTLs, their replication capacity will be compromised. This is indeed found in Japan and in Africa (Nomura et al. 2013; Payne et al. 2014). In Japan, a reduction of replication capacity was observed in subtype B of HIV-1 between 1995 and 2009. Meanwhile, MHC A*24, a common protective MHC type, lost its protective effect in the population. The fitness loss of HIV-1 was associated with mutations in the gag and pol genes. Similarly, viral replication capacity in Botswana was lower than in South Africa because the epidemic in Botswana had been ongoing for a longer period of time, resulting in a more attenuated virus. Interestingly, the average CD4 cell count in treatment-naïve Botswanan patients was lower than in South African patients, i.e., decreased fitness is associated with increased virulence, in agreement with previous analysis of the Nef protein (Liu 2015). The African study also revealed adaptation of HIV-1 to certain MHC types, particularly to the well-known protective types, B*57 and B*58.01.
On the other hand, the North American population is more varied in MHC types. Therefore, when HIV-1 is transmitted into a new host, it is less likely to encounter the same selective pressure as in the previous host. Thus immune escape mutations are more likely to revert in favor of maximum replication capacity. After several decades of interhost evolution in North America, the viral genes have become more diverse, as a result of mutations driven by diverse CTL responses (Cotton et al. 2014, see star-like phylogeny in fig. 5). There are small yet significant increases in the frequency of escape mutations against protective MHC types. However, the study did not find a significant change in replication capacity, although the fitness of modern viruses is distributed over a broader range of values. The ability of the Nef protein to down-regulate CD4 and MHC molecules was gradually optimized to the level of the inferred ancestral virus. It is interesting that the improvement of Nef activities since 1979 is reaching the maximum (notice asymmetrical distribution of dots in Fig. 6 after Cotton et al. 2014, with more dots below the average values than above). Optimization of Nef functions may also account for the increase in viral load levels and decreased CD4 counts in HIV-1 infected patients in the Netherlands (Gras et al. 2009).
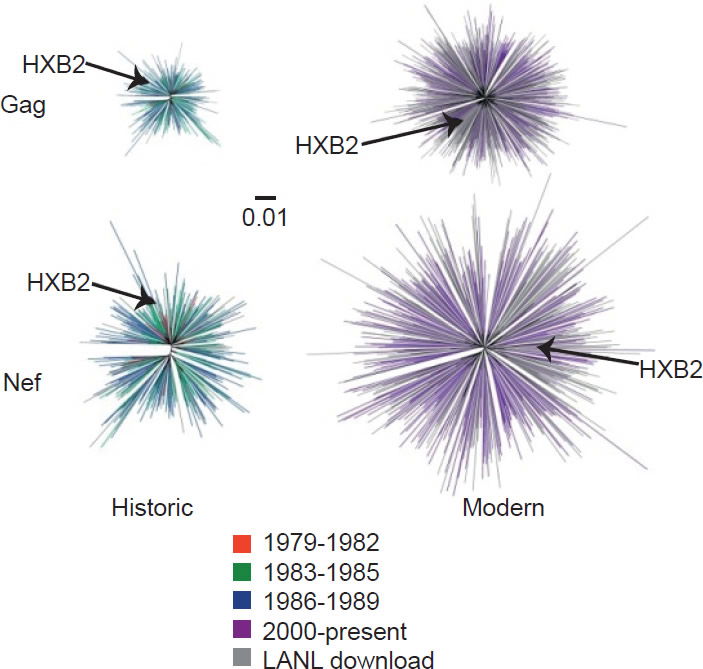
Fig. 5. Diversity of North American Gag and Nef sequences from historic (1979–1989) and modern (2000+) eras, after Fig. 1 of Cotton et al. 2014. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3998893/figure/pgen-1004295-g001/.
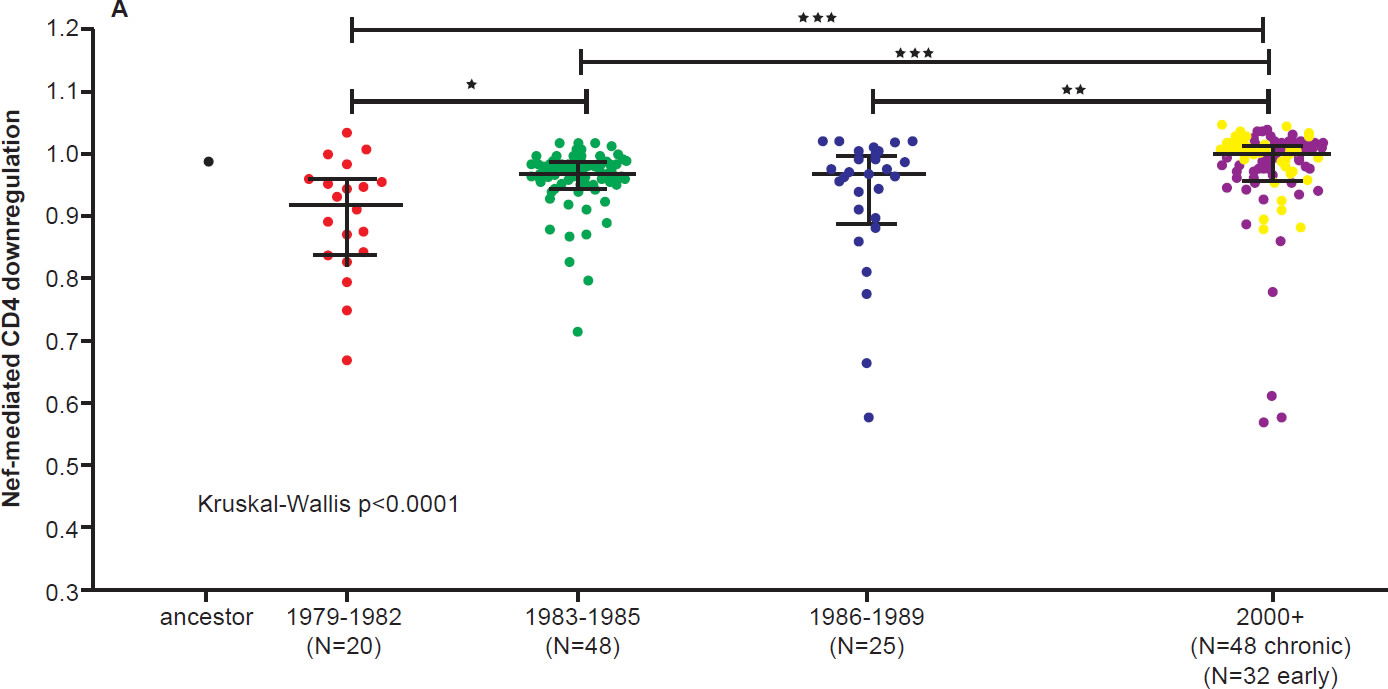
Fig. 6 A. CD4 downregulation activities of the inferred ancestral Nef sequence (mean±S.E.M. of eight replicate measurements) and patient-derived Nef clones from various eras (one per patient, representing the mean of triplicate measurements). CD4 downregulation values are normalized to that of HIV subtype B control Nef strain SF2, such that a value of 1 indicates CD4 downregulation activity equal to that of SF2 while values>1 and <1 indicate activities higher or lower than SF2 respectively. Modern Nefs exhibited significantly higher CD4 downregulation activity compared to historic Nefs (Kruskal-Wallis p<0.0001).
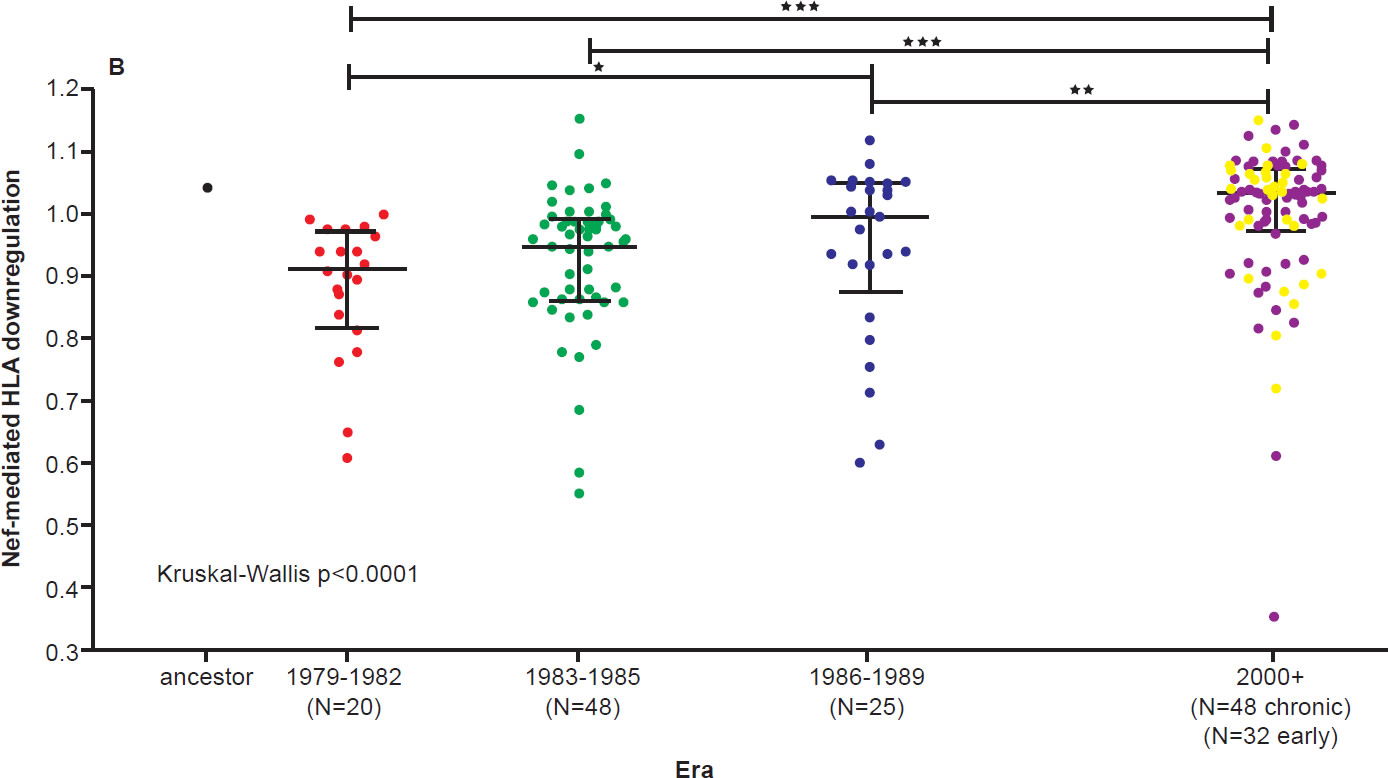
Fig. 6 B. SF2-normalized HLA class I downregulation activities of inferred ancestral (mean±S.E.M. of 8 replicate measurements) and patient-derived Nef sequences (one per patient, mean of triplicate measurements). Modern Nefs exhibited significantly higher HLA downregulation activity compared to historic Nefs (Kruskal-Wallis p<0.0001). After Figs. 8B and 8C of Cotton et al. 2010 at http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3998893/figure/pgen-1004295-g008/.
Random Drift
During the early stages of infection, one founder virus expands exponentially to establish a quasispecies. The process is largely random and subject to genetic drift (Keele et al. 2008). The resulting population provides the basis on which immune selection subsequently acts. Even with strong CTL selection during the first three months of infection, a study by Abrahams et al. (2013) failed to reveal significant differences in the rates of HIV diversification between rapid and slow progressors, indicating much diversification is not attributable to immune selection. Maldarelli et al. (2013) discovered that even non-synonymous mutations may be nearly-neutral and immune to selection.
Pandit and Sinha (2011) analyzed the evolution of codon usage by HIV-1 and compared the codon preferences of the virus and that of the human host. They found the differences in codon preferences between the virus and the host narrowed significantly between the early 1980s and the late 1990s, indicating a period of adaptation between the two species over 15 years. However, the trend changed since then. Codon usage evolution of HIV-1 became stagnant since the 1990s and in fact is drifting away from preferred codon usage of the human species (see fig. 7, after Pandit and Sinha 2011).
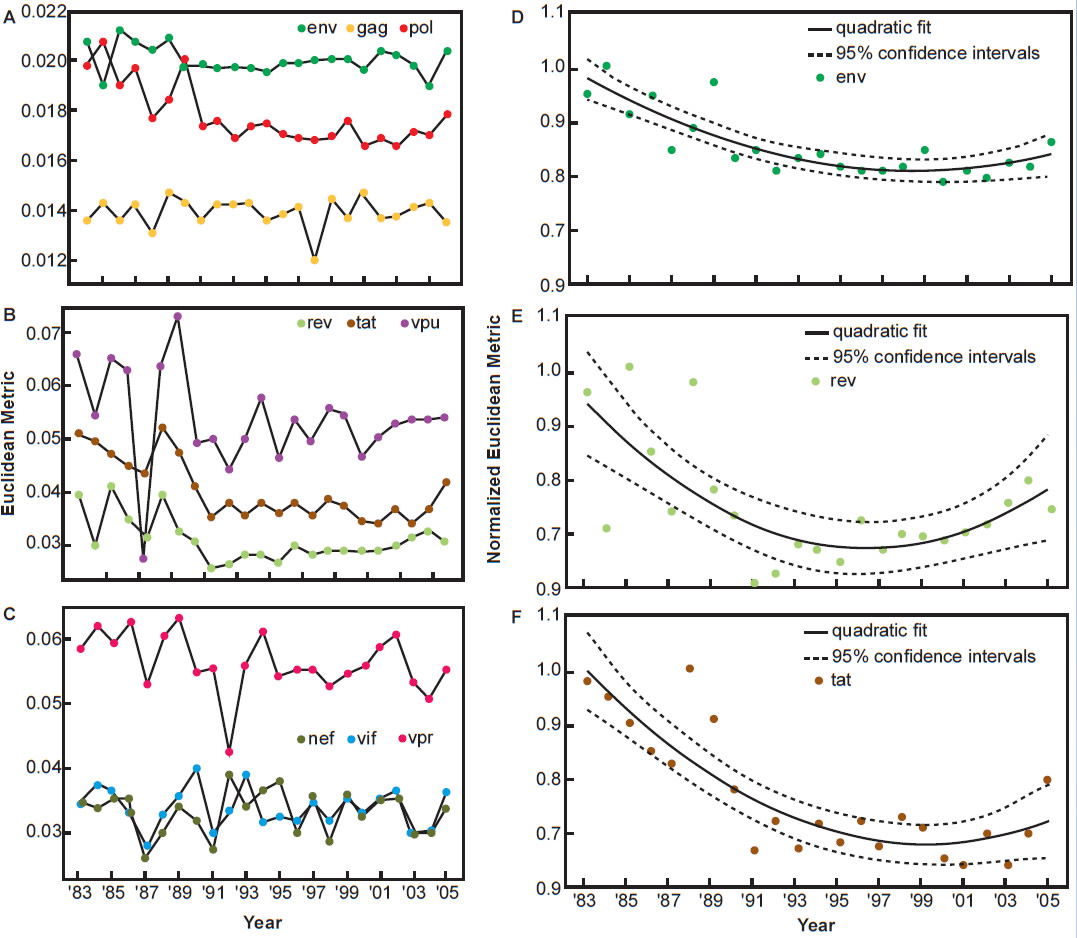
Fig. 7. Temporal variation in the codon usage pattern of HIV-1 genes with respect to human host, after Fig. 5 of Pandit and Sinha, 2011 at http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3245234/figure/pone-0028889-g005/.
The Effect of Antiretroviral Therapy
The above discussion did not consider antiviral treatment. However, in developed countries, antiretroviral therapy has become the primary engine in HIV evolution, driving the virus toward extinction.
Antiretroviral drugs are designed to suppress viral replication, but are frequently resisted by HIV-1 through target mutations. However, drug-resistant mutations are associated with reduced replication capacity, compromised transmissibility, and lower plasma viral load (Machouf et al. 2006; Pingen et al. 2014). For this reason, many clinicians see a value in maintaining sup-optimal therapy when alternative drugs are not available.
There is an estimate that the in vivo mutation rate of HIV-1 is close to its error threshold (Tripathi et al. 2012). Therefore it is feasible to use drugs that increase the mutation rate in reverse transcription of RNA to drive the virus toward error-catastrophe.
How Far Has HIV-1 Degenerated?
Since the AIDS pandemic started three decades ago, the evolution of HIV-1 has been carefully observed and systematically documented. This is especially true for subtype B, the dominant subtype in developed countries. A quick comparison of historical and modern sequences revealed continuous divergence within subtype B. Nucleotide identity decreased from 94.8 ± 0.94% among genomic sequences collected between 1982 and 1985, to 90.0 ± 1.35% among sequences collected between 2011 and 2013. The consensus sequences also differed by about 4%.1 Nucleotide identity between subtypes is 70–90% (See review by Ariën, Vanham, and Arts 2007). If all subtypes of group M indeed descended from a common ancestor transmitted to mankind in the early 1900s, the degeneration rate of HIV-1 would be comparable to that of the H1N1 strain of the influenza virus, which mutated more than 15% of its genome in about a century (see Carter and Sanford 2012. The genome sizes of the two viruses are comparable). While the original H1N1 influenza virus has gone extinct, HIV-1 sill thrives in various forms. This may have to do with the persistent nature of HIV infection. However, in highly prevalent areas of Africa, as well as globally, subtype C, which has a lower replication capacity, is replacing other subtypes due to its relatively higher transmissibility and longer survival of the host (Ariën, Vanham, and Arts 2007).
Nonetheless, HIV-1-associated mortality is still higher in subtype C-dominant areas than in subtype B-dominant areas, presumably because the latter are wealthier countries where antiviral therapies are more accessible (Ortblad, Lozano, and Murray 2013). This indicates that viral attenuation is not the primary cause of the currently observed decline in AIDS mortality.
Conclusion
HIV-1 adapts to host codon preferences, immune pressure, and antiretroviral drugs through mutation. However, most such adaptive mutations carry a fitness cost. Even with diploidy, genome recombination, and persistence in the DNA form, HIV-1 appears to be losing fitness. By coreceptor switch and optimization of viral genes for maximum replication efficiency, the virus may gain virulence, but high virulence compromises its preservation in the human population. While the less virulent subtype C seems to be taking over the epidemic, antiretroviral therapies are driving down the fitness of all subtypes of HIV-1.
References
Abrahams, M-R., F. K .Treurnicht, N. K. Ngandu, S. A. Goodier, J. C. Marais, H. Bredell, R. Thebus et al. 2013. “Rapid, Complex Adaption of Transmitted HIV-1 Full-Length Genomes in Subtype C-infected Individuals with Differing Disease Progression.” AIDS 27 (4): 507–18.
Ariën, K. K., G. Vanham, and E. J. Arts. 2007. “Is HIV-1 Evolving into a Less Virulent Form in Humans?” Nature Reviews Microbiology 5 (2): 141–51.
Arnott, A., D. Jardine, K. Wilson, P. R. Gorry, K. Merlin, P. Grey, M. G. Law et al. 2010. “High Viral Fitness during Acute HIV-1 Infection.” PLoS One 5 (9): e12631.
Behe, M. J. 2007. The Edge of Evolution: The Search for the Limits of Darwinism. New York, New York: Free Press.
Boutwell, C. L., C. F. Rowley, and M. Essex. 2009. “Reduced Viral Replication Capacity of Human Immunodeficiency Virus Type 1 Subtype C Caused By Cytotoxic-T-Lymphocyte Escape Mutations in HLA-B57 Epitopes of Capsid Protein.” Journal of Virology 83 (6): 2460–68.
Carter, R. W., and J. C. Sanford. 2012. “A New Look at an Old Virus: Patterns of Mutation Accumulation in the Human H1N1 Influenza Virus Since 1918.” Theoretical Biology and Medical Modeling 9: 42.
Cotton, L. A., X. T. Kuang, A. Q. Le, J. M. Carlson, B. Chan, D. R. Chopera, C. J. Brumme et al. 2014. “Genotypic and Functional Impact of HIV-1 Adaptation to Its Host Population during the North American Epidemic.” PLoS Genetics 10 (4): e1004295.
Goering, R. V., H. M. Dockrell, M. Zuckerman, I. M. Roitt, and P. L. Chiodini. 2013. Mims’ Medical Microbiology. 5th ed. Philadelphia, Pennsylvania: Elsevier Saunders.
Gras, L., S. Jurriaans, M. Bakker, A. van Sighem, D. Bezemer, C. Fraser, J. Lange, J. M. Prins, B. Berkhout, F. de Wolf, for the ATHENA national observational cohort study. 2009. “Viral Load Levels Measured at Set-Point Have Risen Over the Last Decade of the HIV Epidemic in the Netherlands.” PLoS One 4 (10): e7365.
Kaiser, G. 2008. Doc Kaiser’s microbiology homepage. http://faculty.ccbcmd.edu/courses/bio141/lecguide/unit6/cellular/cmidefense/ctls/u3fg2d.html.
Keele, B. F., E. E. Giorgi, J. F. Salazar-Gonzalez, J. M. Decker, K. T. Pham, M. G. Salazar, C. Sun et al. 2008. “Identification and Characterization of Transmitted and Early Founder Virus Envelopes in Primary HIV-1 Infection.” Proceedings of the National Academy of Sciences USA 105 (21): 7552–57.
Liu, Y. 2015. “Mutations in the nef Gene make HIV-1 More Virulent.” Answers Research Journal 8:333–36. https://answersingenesis.org/biology/disease/mutations-in-nef-gene-make-hiv-1-more-virulent/.
Llano, A., A. Williams, A. Olvera, S. Silva-Arrieta, and C. Brandera. 2013. “Best-Characterized HIV-1 CTL Epitopes: The 2013 Update.” In HIV Molecular Immunology 2013. Edited by K. Yusim, B. Korber, C. Brander, D. Barouch, R. de Boer, B. F. Haynes, R. Koup, J. P. Moore, and B. D. Walker, 3–19. Los Alamos, New Mexico: Theoretical Biology and Biophysics Group. 13-27758.
Machouf, N., R. Thomas, V. K. Nguyen, B. Trottier, M. R. Boulassel, M. A. Wainberg, and J. P. Routy. 2006. “Effects of Drug Resistance on Viral Load in Patients Failing Antiretroviral Therapy.” Journal of Medical Virology 78 (5): 608–13.
Maldarelli, F., M. Kearney, S. Palmer, R. Stephens, J. Mican, M. A. Polis, R. T. Davey et al. 2013. “HIV Populations Are Large and Accumulate High Genetic Diversity in a Nonlinear Fashion.” Journal of Virology 87 (18): 10313–23
Nomura, S., N. Hosoya, Z. L. Brumme, M. A. Brockman, T. Kikuchi, M. Koga, H. Nakamura et al. 2013. “Significant Reductions in Gag-Protease-Mediated HIV-1 Replication Capacity during the Course of the Epidemic in Japan.” Journal of Virology 87 (3): 1465–76.
Ortblad, K. F., R. Lozano, and C. J. Murray. 2013. “The Burden of HIV: Insights from the Global Burden of Disease Study 2010.” AIDS 27 (13): 2003–17.
Pandit, A., and S. Sinha. 2011. “Differential Trends in the Codon Usage Patterns in HIV-1 Genes.” PLoS One 6 (12): e28889.
Payne, R., M. Muenchhoff, J. Mann, H. E. Roberts, P. Matthews, E. Adland, A. Hempenstall et al. 2014. “Impact of HLA-driven HIV Adaptation on Virulence in Populations of High HIV Seroprevalence.” Proceedings of the National Academy of Sciences of the United States of America 111 (50): E5393–E5400.
Pingen, M., R. Sarrami-Forooshani, A. M. J. Wensing, P. van Ham, A. Drewniak, C. A. B. Boucher, T. B. H. Geijtenbeek, and M. Nijhuis. 2014. “Diminished Transmission of Drug Resistant HIV-1 Variants with Reduced Replication Capacity in a Human Transmission Model.” Retrovirology 11: 113.
Salgado, M., T. P. Brennan, K. A. O’Connell, J. R. Bailey, S. C. Ray, R. F. Siliciano, and J. N. Blankson. 2010. “Evolution of the HIV-1 nef Gene in HLA-B*57 Positive Elite Suppressors.” Retrovirology 7: 94.
Sanford, J. C. 2014. Genetic Entropy. Geneva, New York: FMS Publications.
Sharp, P. M., and B. H. Hahn. 2011. “Origins of HIV and the AIDS Pandemic.” Cold Spring Harbor Perspectives in Medicine 1 (1): a006841
Shriner, D., R. Shankarappa, M. A. Jensen, D. C. Nickle, J. E. Mittler, J. B. Margolick, and J. I. Mullins. 2004. “Influence of Random Genetic Drift on Human Immunodeficiency Virus Type 1 env Evolution During Chronic Infection.” Genetics 166 (3): 1155–64.
Tripathi, K., R. Balagam, N. K. Vishnoi, and N. M. Dixit. 2012. “Stochastic Simulations Suggest that HIV-1 Survives Close to Its Error Threshold.” PLoS Computational Biology 8 (9): e1002684.
Troyer, R. M., K. R. Collins, A. Abraha, E. Fraundorf, D. M. Moore, R. W. Krizan, Z. Toossi et al. 2005. “Changes in Human Immunodeficiency Virus Type 1 Fitness and Genetic Diversity during Disease Progression.” Journal of Virology 79 (14): 9006–18.
Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez et al. 2003. “Antibody Neutralization and Escape by HIV-1.” Nature 422 (6929): 307–12.
Wertheim, J. O., and M. Worobey. 2009. “Dating the Age of the SIV Lineages That Gave Rise to HIV-1 and HIV-2.” PLoS Computational Biology 5 (5): e1000377.
Yue, L., K. J. Pfafferott, J. Baalwa, K. Conrod, C. C. Dong, C. Chui, R. Rong et al. 2015. “Transmitted Virus Fitness and Host T Cell Responses Collectively Define Divergent Infection Outcomes in Two HIV-1 Recipients.” PLoS Pathogens 11 (1): e1004565.