The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
In 2013, it was shown that the alleged interstitial telomeric repeat site of the human chromosome 2 fusion corresponding to chimpanzee chromosomes 2A and 2B of a hypothetical common ancestor was actually a second promoter in the DDX11L2 long noncoding RNA gene. Additional ENCODE related data are provided in this report that not only debunk evolutionary criticism and obfuscation in response to this discovery, but solidify the original finding. New data come from epigenetic-modifications, transcription factor binding, and transcription start site information. It is also shown that the alleged cryptic centromere site, which is very short in length compared to a normal centromere, is completely situated inside the actively expressed protein coding gene ANKRD30BL—encoding both exon and intron regions. Other factors refuting this region as a cryptic centromere are also discussed. Taken together, genomic data for both the alleged fusion and cryptic centromere sites refute the concept of fusion in a human-chimpanzee common ancestor.
Keywords: chromosome 2 fusion, DDX11L2 gene, human evolution, human-chimpanzee, cryptic centromere
Introduction
A major argument supposedly providing evidence of human evolution from a common ancestor with chimpanzees is the “chromosome 2 fusion model” in which ape chromosomes 2A and 2B purportedly fused end-to-end, forming human chromosome 2 (Ijdo et al. 1991; Yunis and Prakash 1982). This idea is postulated despite the fact that all known fusions in extant mammals involve satellite DNA and breaks at or near centromeres. Only telomere-satelliteDNA or satelliteDNA-satelliteDNA are found in chromosomal fusion sites of living animals in nature, not telomere-telomere fusions (Adega, Guedes-Pinto, and Chaves 2009; Chaves et al. 2003; Tsipouri et al. 2008). While telomere-telomere fusions have been documented in the rearranged genomes of human cancer cells, these are not indicative of healthy cells but instead are associated with genomic instability (Tanaka, Beam, and Caruana 2014; Tanaka et al. 2012; Tu et al. 2015). See Fig. 1 for a graphical depiction of the alleged fusion.
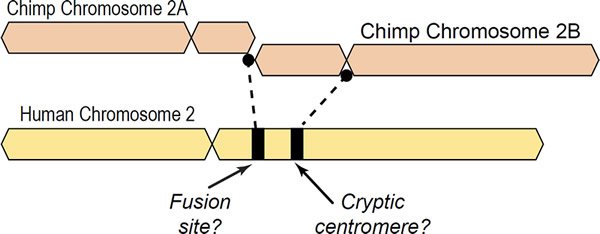
Fig. 1. Depiction of a hypothetical scenario in which chimpanzee chromosomes 2A and 2B fused end-to-end to form human chromosome 2. All chromosomes were comparatively drawn to scale according to cytogenetic images by Yunis and Prakash (1982). Based on cytogenetic scale, there is about 10% of chimpanzee DNA unaccounted for in the alleged fusion.
Another significant aspect questioning the veracity of the fusion site is the fact that it is very small and highly degenerate. Typical human telomeres are 5000 to 15,000 bases in length (Tomkins and Bergman 2011). An end-to-end fusion as proposed by evolutionists would give a signature of at least 10,000 bases in length, yet the fusion site is only 798 bases in length. Furthermore, given the supposed 3 to 6 million years of divergence from a common ancestor (Fan et al. 2002) the fusion site is only 70% identical in sequence compared to a pristine fusion sequence of the same size.
However, the strongest evidence for negation of fusion was published in 2013 in which it was shown that the purported fusion site (read in the minus strand orientation) is a functional DNA binding domain inside the first intron of the DDX11L2 noncoding RNA helicase gene acting as a second promoter (Tomkins 2013). See Fig. 2 for a simplified graphic of the DDX11L2 gene and the alleged fusion site within it. Specifically, as will be shown in this paper, the fusion site sequence binds to at least 12 different transcription factors, including RNA polymerase II, the key enzyme that transcribes genes. Additional data in this paper will also show that along with the binding of RNA polymerase is the fact that transcription has also been shown to initiate inside the fusion-like sequence in a classic promoter-like fashion. These data also intersect with transcriptionally active histone marks and open active chromatin profiles that are hallmarks of promoters. These data, as a whole, strongly validate the alleged fusion sequence as a functional promoter element, not some random accident of fusion.
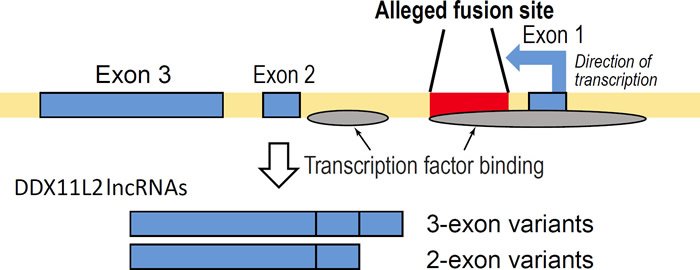
Fig. 2. Simplified graphic showing the fusion site inside the DDX11L2 gene illustrating the two sets of short and long transcript variants produced along with areas of transcription factor binding. Arrow in first exon depicts direction of transcription.
The DDX11L2 long noncoding RNA gene containing the alleged fusion sequence encodes several transcript variants expressed in at least 255 different cell and/or tissue types (Tomkins 2013). The DDX11L2 gene produces RNA transcripts of two different lengths—short variants (~1700 bases long) and long variants (~2200 bases long). In this respect, the fusion site itself appears to be the functional start site for the shorter transcript variants (fig. 1). Furthermore, annotated DDX11L2 gene transcripts suggest complex post-transcriptional regulation through a variety of microRNA binding sites (Tomkins 2013). Many of these microRNA binding sites are shared with the DDX11 protein coding gene transcripts (Tomkins 2013). In fact, both the DDX11L2 and DDX11 genes are significantly coexpressed together in the same tissues (Tomkins 2013). Shared microRNA binding domains and coexpression putatively suggest co-regulation between a protein coding gene and its noncoding RNA pseudogene counterpart as depicted in the classic case of PTEN and PTEN pseudogene transcription (Johnsson et al. 2013).
In summary, chromosome fusions would not be expected to form complexly transcribed and regulated multi-exon, alternatively spliced functional genes. The clear genetic evidence strongly refutes the claim that human chromosome 2 is the result of an ancestral telomeric end-to-end fusion.
Needless to say, such a discovery has caused quite a stir in the evolutionary community. However, the central facts surrounding these recent discoveries negating fusion remain to be challenged since they are now well-documented features of the human genome. Nevertheless, a number of attempted rebuttals of my research posted on various internet blogs have appeared. While none of these efforts have disproven the central facts surrounding fusion negation, they do attempt to either downplay or question aspects of the scenario or mold them to fit the evolutionary model. Each of these so-called arguments attempting to downplay the significance of the data for fusion negation are addressed in turn.
Cryptic Fusion Site Positioning
One internet post put up by a computer scientist (now removed) attempted to question my graphical depiction of the chromosome fusion (Tomkins 2013) which was drawn to scale based on the actual cytogenetic images of Yunis and Prakash (1982) and presented again here in Fig. 1. The actual method that I used was an inconvenient fact that the author of the post deceptively omitted, making it seem as if I produced the images without any source of reference. He then attempted to locate the fusion site using the current versions of the human and chimpanzee genomes which on the surface would seem like a reasonable thing to do except for the problem that the chimpanzee genome is still an unfinished draft sequence and contains numerous gaps filled with meaningless “N”s instead of nucleotides, which do not correspond to the actual length of the unknown gap sizes.
The critic’s method of computationally reconstructing the fusion site location in a graphical image is based on the characters of A, T, C, G, and N in the FASTA files of chimpanzee chromosomes 2A and 2B. Thus, this method is highly susceptible to a garbage in garbage out scenario. In light of this fact, this author set out to ascertain the actual amount of N contained in chimpanzee chromosomes 2A and 2B (panTro4).
For chr2A the total length is 113,622,374 bases of which 7.2% are Ns (8,216,535 in total) corresponding to 5575 total gaps with 3762 gaps being greater than 10 Ns in length. The average (mean) gap size was 1473 Ns with the largest one being 3,000,000. For chr2B the total length is 247,518,478 bases of which 48.6% are Ns (120,253,698 in total) corresponding to 5944 gaps with 3893 being greater than 10 Ns in length with the largest one being 114,098,230. However, it is hardly fair to report an average gap size for chr2B because the largest gap is deliberately set to a high value. As noted by biologist Robert Carter (Creation Ministries International, pers. comm.), the entire first half of the chromosome is a long series of Ns in a deliberate alignment to human to allow for easy evolutionary comparisons to human chromosome 2. Carter also notes that the chromosome is flipped, with the q arm first, for the same reason.
These data indicate that especially chimpanzee chromosome 2B is bloated with Ns comprising nearly half its alleged length. Even chromosome 2A contains over 7% Ns. Clearly the addition of these huge numbers of meaningless Ns indicates that these chromosomes are literally bloated with worthless data for reconstructing the specific location of the fusion site or the alleged cryptic centromere site (discussed below). In fact, this author verified the fact that the number of Ns in an assembly gap do not correspond with actual gap size through personal correspondence with NCBI curatorial staff.
Due to the fragmented nature of chimpanzee chromosomes 2A and 2B that are bloated with meaningless Ns and aligned onto human as a reference, a better method based on drawn-to-scale cytogenetic images is the best that can be achieved at this point in time. This is what I based my original image on which showed that an approximate 10% loss of chimpanzee chromosome 2B would have occurred in a fusion and that the alleged fusion site is not located where it is usually depicted.
Chromosome 2 Fusion—What Should We Expect?
As stated above in the introduction, the general model proposed by evolutionary scientists is that the chromosome 2 fusion event was a head-to-head telomeric fusion. However, because the alleged fusion site is so small and degenerate, evolutionists in general propose that somehow the telomere repeats degenerated. Some evolutionists have proposed that the telomeres broke off prior to fusion and that the fusion site may actually be the product of a head-to-head subtelomere fusion (Rudd 2014).
Subtelomeres are intervening regions between the telomere repeats and the internal regions of chromosomes. They are enriched with degenerate telomere repeats (similar to the fusion site), protein coding genes, noncoding RNA genes, segmental duplications, and the same type of repetitive elements found elsewhere throughout the genome as opposed to telomeres that only contain near-perfect TTAGGG repeats (Riethman et al. 2004). On the surface, a fusion of subtelomeres would seem to resolve some of the problems inherent to the fusion hypothesis. However, in reality, bringing up the issue of subtelomeres in relation to fusion actually compounds the overall problem.
While human telomeres are 5000 to 15, 000 bases in length, subtelomere regions on average are 100,000 to 300,000 bases in length and comprise up to 5% of the human genome (Martin et al. 2002; Riethman et al. 2004). When we include the amount of DNA that should be present in a signature of subtelomeres, the problem becomes even more problematic given that the fusion site is only 798 bases in length. One evolutionary response to this argument could be that the fusion occurred just barely beyond the centromere proximal ends of each subtelomere. But this reasoning is problematic too since many critical genes needed for life exist in subtelomeres and their gene density is the same as the rest of the genome (Riethman et al. 2004). While subtelomeres appear to be designed as regions of variability to confer phenotypic diversity associated with an increase in recombination hotspots and segmental duplications, major deletions in these regions are detrimental in humans and associated with a variety of developmental abnormalities and diseases (Lemmers et al. 2010; Linardopoulou et al. 2005, 2007). If the alleged 798-base fusion site represents a real head-to-head fusion of shortened/ truncated subtelomeres, then a major loss of about 200,000 or more bases of gene rich DNA would have occurred.
But the fusion becomes even more untenable when we consider the complete absence of chimpanzee subtelomeric specific heterochromatic repeats that are missing in and around the alleged fusion signature that should have been present at some level in the hypothetical hominid common ancestor (Ventura et al. 2012). As stated in the publication by Ventura et al., “Chimpanzee and gorilla chromosomes differ from human chromosomes by the presence of large blocks of subterminal heterochromatin thought to be composed primarily of arrays of tandem satellite sequence.” In fact, the amount of chimpanzee subtelomeric sequence now mysteriously missing in humans is amazingly large. Ventura et al. state, “While variable in size, most of the satellite tracts were >20 kbp, with some exceeding 60 kbp within a given BAC [bacterial artificial chromosome—a cloned large insert genomic fragment in a single copy bacterial vector].” The presence of abundant amounts of chimpanzee-specific subtelomeric satellite DNA sequence in chimpanzee, but completely absent in human, is not only a major problem for the chromosome 2 fusion model, but human evolution from a common ancestor with chimpanzee in general. This is because major changes must have occurred in humans in a “short” time frame of only 3 to 6 mya.
Another major problem with either the subtelomeric or telomeric fusion models for human chromosome 2 is that no homology exists for the fusion region to the respective ends of chimpanzee chromosomes 2A and 2B. In the Ventura et al. (2012) paper, they attempted to hybridize the fusion site region to chimpanzee chromosomes and reported, “We observed no signals to the chimpanzee subcap locations using fosmid clones from the chromosome 2 region.” Tomkins and Bergman had reported the same results using a BLASTN analysis in 2011, although these results would not have been conclusive since chimpanzee subtelomeres are not well sequenced. The Ventura et al. (2012) study using fluorescent in situ hybridization, however, confirms the fact that no evolutionary hallmarks of fusion exist in a human-chimpanzee common ancestor believed to have occurred after the human-chimpanzee divergence (Ventura et al. 2012).
Significance of the Alleged Fusion Site as a Gene Promoter
Perhaps the greatest problem for the alleged fusion site is its functionality as a second promoter in the DDX11L2 noncoding RNA gene as demonstrated by transcription factor binding, RNA polymerase 2 binding, and promoter-based epigenetic profiles for several different types of histone modifications as initially presented by Tomkins in 2013.
A number of internet postings have attempted to downplay the significance of the well documented promoter activity at the alleged fusion site by comparing the activity of the noncoding DDX11L2 gene to that of protein coding genes. However, one thing that is important to note is that long noncoding RNA (lncRNA) genes cannot be properly compared to protein coding genes because lncRNA genes are expressed at much lower levels (Kornienko et al. 2016). Thus, their biological significance cannot be measured simply by comparing them to protein coding genes—an important detail that ill-informed critics don’t realize. Combinatorial data have to be compiled including epigenetic marks, transcription factor binding, transcription start site data, expression levels, and co-expression data with other genes.
Over 56,000 lncRNA sites across the genome have been described in a variety of human cell types and new lncRNA genes continue to be increasingly identified as new tissues and cell types are studied (Iyer et al. 2015; Xie et al. 2013). At the beginning of their discovery, lncRNAs were initially characterized as regulators of chromatin in the nucleus (Khalil et al. 2009). But as increasing numbers of lncRNAs are analyzed, it has become evident that they play essential roles in a wide variety of cellular processes (Quinn and Chang 2016).
A comprehensive inspection of the UCSC genome browser data for transcription factor binding shows that over 80 transcription factors bind to the DDX11L2 gene in and around its promoter regions (fig. 3). In the alleged fusion region, the second promoter, at least 12 transcription factors have been shown to bind (figs. 3 and 4). These binding sites overlap with tracks that are highly significant for transcriptionally active histone modifications. To further confirm the transcriptional activity at the alleged fusion site, the FANTOM4, FANTOM5, and ENCODE databases (fantom.gsc.riken.jp) were queried for transcription start site data based on cap analysis of gene expression (CAGE) data in which RNA transcripts sequenced from the five prime end are aligned onto their respective genomic promoter sequence. As shown in Fig. 5, all three databases showed transcript initiation within the 798 base alleged fusion site sequence in the classic transcription start site cluster signature for a gene promoter (Haberle et al. 2015).
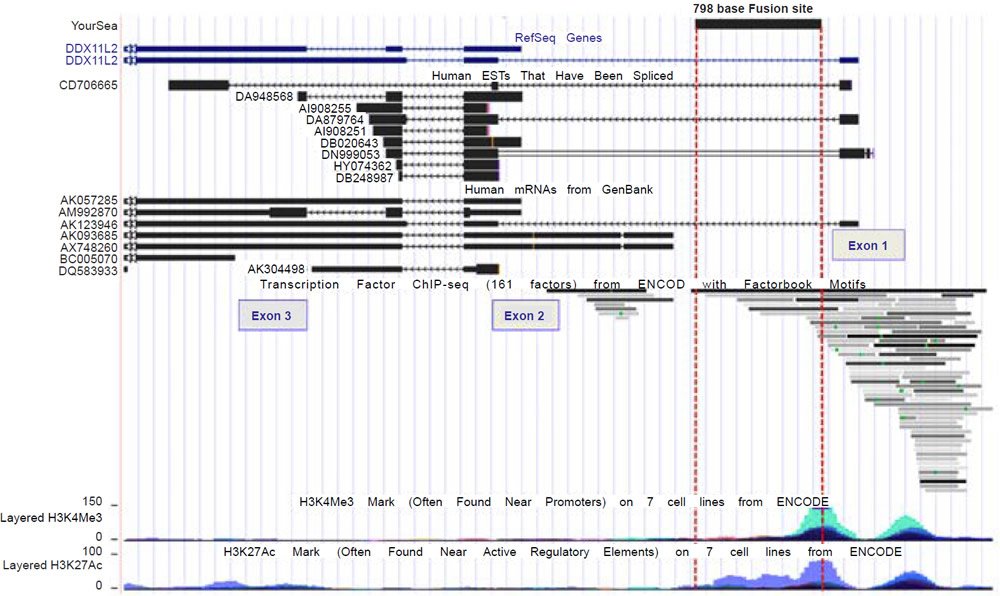
Fig. 3. UCSC genome browser view of the DDX11L2 gene, its aligned mRNAs, transcription factor binding, and transcriptionally active histone tracks in relation to the alleged fusion site.

Fig. 4. A close-up view of the transcription factor binding tracks within the 798 base alleged fusion site.
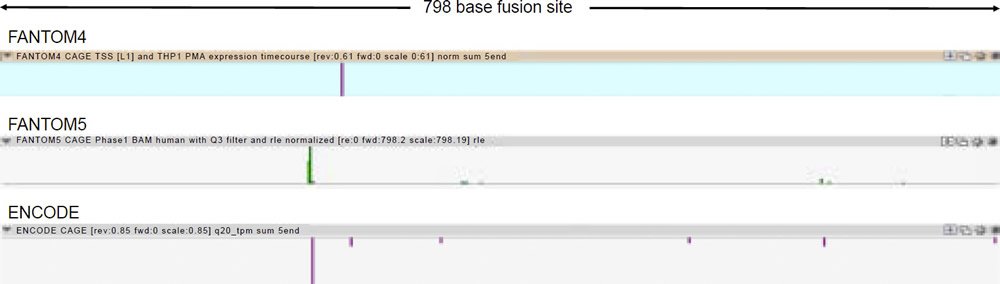
Fig. 5. Transcription start site data from FANTOM4, FANTOM5, and ENCODE cap analysis of gene expression (CAGE) data for Chr2:114,360,257-114,361,054 (http://fantom.gsc.riken.jp).
All of the combinatorial genomic data clearly show that the alleged fusion site is a second promoter in the DDX11L2 gene. In addition, the transcriptional activity at the alleged fusion site is neither nominal nor spurious.
Ken Miller Errors on Fusion
One of the main proponents of the alleged fusion site as evidence of human evolution among the general public is Ken Miller at Brown University. Miller initially claimed that I was wrong about the fusion site being situated inside the first intron of the DDX11L2 gene based on data that excluded the three-exon version of the gene (Mooney 2014). I then conversed with Miller via email and he did concede that when one includes the longer transcript variants, the fusion site is in fact located inside the gene (Luskin 2014).
Following these exchanges with Miller, he attempted to explain the longer transcripts of the DDX11L2 gene, by claiming that they “are easily explained by variability in the termination of transcription so that occasionally a somewhat longer RNA is produced” (Moran 2015). This argument is completely erroneous because the direction of transcription for the DDX11L2 gene which is encoded on the minus strand is the opposite of what Miller is claiming. The longer transcripts begin ahead of the fusion site at the five prime end of the gene. It is a well documented fact that transcription begins at the five prime ends of genes.
As depicted in Fig. 2, the DDX11L2 gene is located on the minus strand and termination occurs at the opposite end of the gene from which Miller is claiming. This completely erroneous argument is Miller’s chief complaint concerning the multiple facts refuting fusion.
Miller and others have also promoted the idea that transcription factor binding activity in the alleged fusion site is insignificant or spurious. However, as noted above, the data for promoter functionality of the alleged fusion site is combinatorial and conclusive.
The Cryptic Centromere Site—Not So Centromeric After All
Centromeres are specific regions in chromosomes defined by both sequence and epigenetic profiles that play a physical role in the assembly of the kinetochore—a complex structure involving multiple proteins that provides a central function in chromosome segregation during cell division. In a chromosome fusion, two centromeres would initially exist and one would have to be deactivated to maintain proper cellular function. It is postulated that the inactivated centromere would eventually degrade and become a cryptic genomic fossil.
A very recent scientific paper has been published that would seem to bolster the evidence for a cryptic centromere associated with the chromosome 2 fusion event (Miga 2016). The paper places the main argument for a fusion event and a cryptic centromere on gene synteny with corresponding hypothetical fusion-related areas in the chimpanzee genome surrounding the alleged cryptic centromere site. Note that the synteny is emphasized, not the specific genomic location because as noted previously, over 48% of chimpanzee chr2B which allegedly contributed the cryptic centromere is nothing but meaningless N in thousands of gaps with one large notable gap covering nearly half the chromosome. Clearly the assembly of chimpanzee chromosome 2B is full of meaningless data. In other words, proclaiming that gene synteny supports fusion based on a highly suspect assembly using human as a scaffold is an error of logic called begging the question. This is a fallacious argument where the conclusion is assumed in one of the premises. At this point, the issue of synteny between human and chimpanzee around the cryptic centromere remains to be resolved until an unbiased assembly of the chimpanzee genome is achieved.
Another serious problem with the alleged cryptic centromere is that its human alphoid repeat DNA sequence does not closely match homologous chimpanzee centromeres and chromosomes (Archidiacono et al. 1995). Species specificity of human centromeres was originally documented using fluorescent in situ hybridization of 27 different human alphoid repeats in chromosomal spreads of chimpanzees and gorilla (Archidiacono et al. 1995). The authors of the paper state, “The surprising results showed that the vast majority of the probes did not recognize their corresponding homologous chromosomes.” They also stated that the experiments “yielded very heterogeneous results: some probes gave intense signals, but always on nonhomologous chromosomes; others did not produce any hybridization signal.” In fact the differences in human vs. chimpanzee alphoid repeat arrays are so profound that Haaf and Willard in a later publication stated, “This implies that the human-chimpanzee sequence divergence has not arisen from a common ancestral α-satellite repeat(s)” (Haaf and Willard 1997).
To test this idea of nonhomology from a bioinformatics perspective, I used the online version of the BLASTN algorithm with gap extension and no repeat masking to query the 171 base human consensus alphoid sequence (accession X07685) against both the human and chimpanzee genomes (figs. 6a and 6b). In confirmation of the species specificity found in fluorescent in situ hybridization studies, much fewer hits occurred on the chimpanzee genome than human. Also in contrast to human where a large number of chromosomes had interstitial alphoid regions outside the centromere, most hits occurred at or near centromeres, with three chimpanzee chromosomes having no hits. Of particular interest to the idea of a cryptic centromere is the fact that human chromosome 2 had six internal regions bearing alphoid repeats outside the centromere. Clearly the alphoid region at the alleged cryptic centromere site is not a unique or an uncommon feature in the human genome outside centromeres, especially in chromosome 2. Thirteen other human chromosomes also showed multiple regions of alphoid repeats outside the centromere.
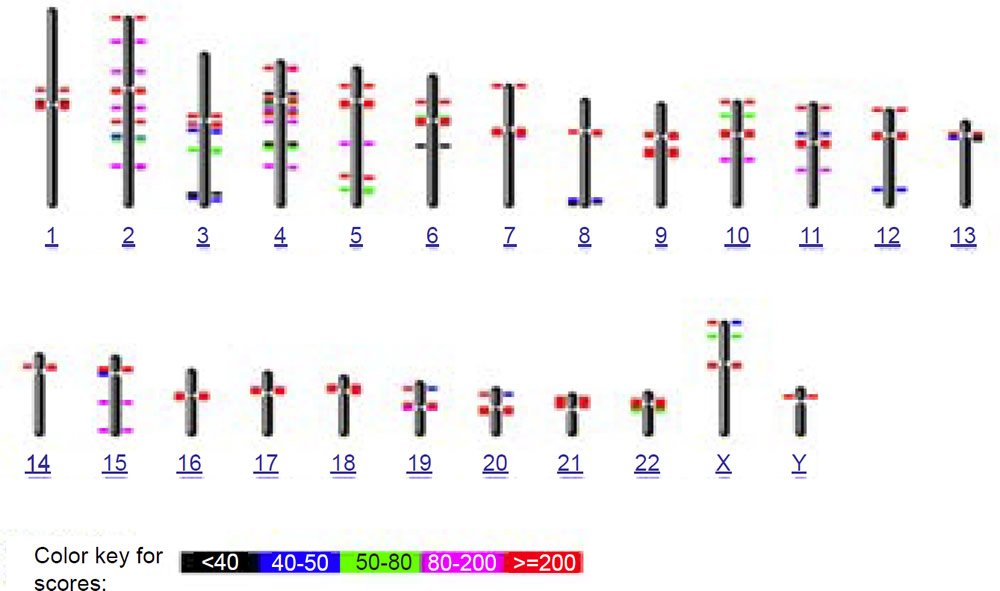
(a) BLASTN hits for 171 base consensus alphoid repeat on human
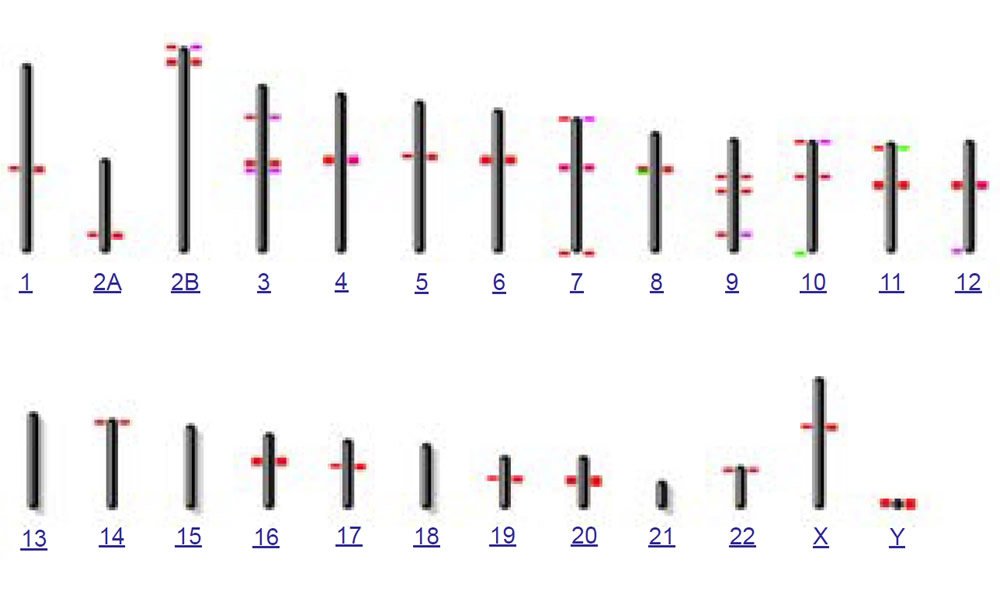
(b) BLASTN hits for 171 base consensus alphoid repeat on chimp
Fig. 6. BLASTN results from querying the 171 base human consensus alphoid sequence (accession X07685) against both the (a) human (hg19) and (b) chimpanzee (PanTro4) genomes using gap extension and no repeat masking.
Another problem with the alleged cryptic centromere is its short length. The cryptic centromere site is extremely small compared to a real centromere—it is only 41,608 bases in length, but this also includes two insertions of a LPA3/LINE repeat (5957 bases) and a SVA-E element (2571 bases) (fig. 7). Subtracting the insertions of these non-alphoid elements gives a length of only 33,080 bases—a fraction of the size of human centromeres that range in length between 250,000 and 5,000,000 bases (Aldrup-Macdonald and Sullivan 2014). Thus, if this was in fact a relic centromere of an ancient chromosome fusion, its size should be greater than six times its current length at the minimum.
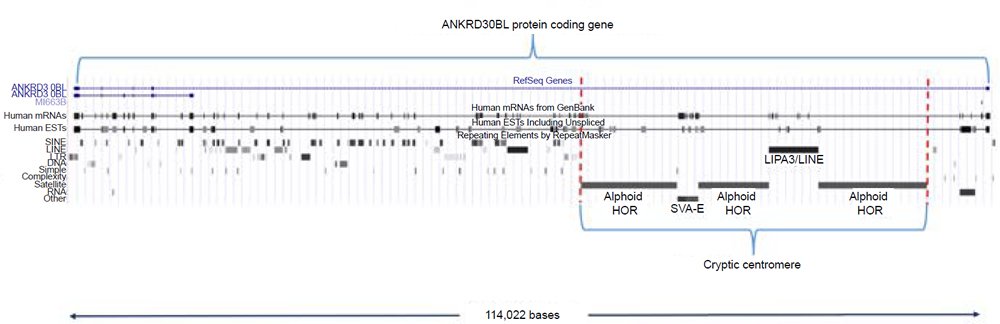
Fig. 7. The 41,608 base cryptic centromere region situated within the ANKRD30BL protein coding gene. UCSC coordinates for the entire gene region are chr2:132,146,999-132,261,000 (hg19).
Perhaps the greatest problem with the idea of a cryptic centromere is that it is completely situated inside the actively expressed protein coding gene ANKRD30BL [Ankyrin Repeat Domain 30B Like] with sections of it located in both intron and exon regions according to aligned transcripts (fig. 7). Ankyrin is a specific protein associated with the plasma membrane in eukaryotic cells and is involved in the interaction of the cytoskeleton with integral membrane proteins. Proteins with ankyrin repeats such as ANKRD30BL are called ankyrin-like proteins. The ankyrin repeat of 33 amino acids has been implicated in embryogenesis and the regulation of many intracellular processes based on protein-protein interactions (Voronin and Kiseleva 2008). The fact that the cryptic centromere is a functional region inside a protein coding gene encompassing both protein coding and noncoding regions strongly implies that it is a key gene feature, not a defunct centromere.
Summary
Since my original 2013 publication, the data invalidating the chromosome 2 fusion hypothesis has become even more compelling with the addition of more ENCODE and FANTOM database information in regard to the alleged fusion site. At present, at least 86 different transcription factors bind to the DDX11L2 gene in and around its two promoter regions with 12 transcription factors binding in the alleged fusion site (including RNA polymerase 2). These binding sites coincide with highly significant levels of transcriptionally active histone modifications. In further confirmation of the transcriptional activity at the alleged fusion site, the FANTOM4, FANTOM5, and ENCODE databases all show transcript initiation within the 798 base alleged fusion site sequence in the classic signature for a transcription start site cluster associated with a gene promoter. All of these combinatorial genomic data clearly show that the alleged fusion site is a promoter in the DDX11L2 gene and that the activity is neither nominal or spurious.
Additional data presented in this report also shows that the alleged cryptic centromere site has no homologous chromosomal matches in the chimpanzee genome. It is also shown the site is too small to represent a centromere and that interstitial alphoid repeat regions are common on many human chromosomes including chromosome 2 that has six different alphoid-rich repeat regions outside the centromere. However, the most negating evidence against the cryptic centromere site is that it is completely situated inside the actively expressed protein coding gene ANKRD30BL [Ankyrin Repeat Domain 30B Like]. In fact, sections of it extend across both intron and exon regions according to aligned transcripts. The alleged cryptic centromere is clearly a functional part of a protein coding gene and not the remnant of a centromere.
References
Adega, F., H. Guedes-Pinto, and R. Chaves. 2009. “Satellite DNA in the Karyotype Evolution of Domestic Animals— Clinical Considerations.” Cytogenet Genome Research 126 (1–2): 12–20. doi:10.1159/000245903.
Aldrup-Macdonald, M. E., and B. A. Sullivan. 2014. “The Past, Present, and Future of Human Centromere Genomics.” Genes (Basel) 5 (1): 33–50.
Archidiacono, N., R. Antonacci, R. Marzella, P. Finelli, A. Lonoce, and M. Rocchi. 1995. “Comparative Mapping of Human Alphoid Sequences in Great Apes Using Fluorescence in situ Hybridization.” Genomics 25 (2): 477–484.
Chaves, R., F. Adega, J. Wienberg, H. Guedes-Pinto, and J. S. Heslop-Harrison. 2003. “Molecular Cytogenetic Analysis and Centromeric Satellite Organization of a Novel 8;11 Translocation in Sheep: A Possible Intermediate in Biarmed Chromosome Evolution.” Mammalian Genome 14 (10): 706–710.
Fan, Y., E. Linardopoulou, C. Friedman, E. Williams, and B. J. Trask. 2002. “Genomic Structure and Evolution of the Ancestral Chromosome Fusion Site in 2q13-2q14.1 and Paralogous Regions on other Human Chromosomes.” Genome Research 12 (11): 1651–1662. doi:10.1101/gr.337602.
Haaf, T., and H. F. Willard. 1997. “Chromosome-Specific Alpha-Satellite DNA from the Centromere of Chimpanzee Chromosome 4.” Chromosoma 106 (4): 226–232.
Haberle, V., A. R. R. Forrest, Y. Hayashizaki, P. Carninci, and B. Lenard. 2015. “CAGEr: Precise TSS Data Retrieval and High-Resolution Promoterome Mining for Integrative Analyses.” Nucleic Acids Research 43 (8): e51. doi:10.1093/nar/gkv054.
Ijdo, J. W., A. Baldini, D. C. Ward, S. T. Reeders, and R. A. Wells. 1991. “Origin of Human Chromosome 2: An Ancestral Telomere-Telomere Fusion.” Proceedings of the National Academy of Sciences USA 88 (20): 9051–9055.
Iyer, M. K., Y. S. Niknafs, R. Malik, U. Singhal, A. Sahu, Y. Hosono, T. R. Barrett, et al. 2015. “The Landscape of Long Noncoding RNAs in the Human Transcriptome.” Nature Genetics 47 (3): 199–208. doi:10.1038/ng.3192.
Johnsson, P., A. Ackley, L. Vidarsdottir, W.-O. Lui, M. Corcoran, D. Grandér and K. V. Morris. 2013. “A Pseudogene Long-Noncoding-RNA Network Regulates PTEN Transcription and Translation in Human Cells.” Nature Structural & Molecular Biology 20 (4): 440–446. doi:10.1038/nsmb.2516.
Khalil, A. M., M. Guttman, M. Huarte, M. Garber, A. Raj, M. D. Rivea, K. Thomas, et al. 2009. “Many Human Large Intergenic Noncoding RNAs Associate with Chromatin- Modifying Complexes and Affect Gene Expression.” Proceedings of the National Academy of Sciences USA 106 (28): 11667–11672. doi:10.1073/pnas.0904715106.
Kornienko, A. E., C. P. Dotter, P. M. Guenzl, H. Gisslinger, B. Gisslinger, C. Cleary, R. Kralovics, F. M. Pauler, and D. P. Barlow. 2016. “Long Non-Coding RNAs Display Higher Natural Expression Variation than Protein-Coding Genes in Healthy Humans.” Genome Biology 17: 14. doi:10.1186/s13059-016-0873-8.
Lemmers, R. J., P. J. van der Vliet, R. Klooster, S. Sacconi, P. Camaño, J. G. Dauwerse, L. Snider, et al. 2010. “A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy.” Science 329 (5999): 1650–1653. doi:10.1126/science.1189044.
Linardopoulou, E. V., S. S. Parghi, C. Friedman, G. E. Osborn, S. M. Parkhurst, and B. J. Trask. 2007. “Human Subtelomeric WASH Genes Encode a New Subclass of the WASP Family.” PLoS Genetics 3 (12): e237. doi:10.1371/journal.pgen.0030237.
Linardopoulou, E. V., E. M. Williams, Y. Fan, C. Friedman, J. M. Young, and B. J. Trask. 2005. “Human Subtelomeres are Hot Spots of Interchromosomal Recombination and Segmental Duplication.” Nature 437 (7055): 94–100. doi:10.1038/nature04029.
Luskin, C. 2014. “Defenders of the Evolutionary ‘Consensus’ could Benefit from more Fact Checking.” The Blaze, August 27, 2014. http://www.theblaze.com/contributions/defenders-of-the-evolutionary-consensus-could-benefit-from-morefact-checking-2/.
Martin, C. L., A. Wong, A. Gross, J. Chung, J. A. Fantes, and D. H. Leadbetter. 2002. “The Evolutionary Origin of Human Subtelomeric Homologies—Or Where the Ends Begin.” American Journal of Human Genetics 70 (4): 972–984. doi:10.1086/339768.
Miga, K. H. 2016. “Chromosome-Specific Centromere Sequences Provide an Estimate of the Ancestral Chromosome 2 Fusion Event in Hominin Genomes.” Journal of Heredity 108 (1): 45–52. doi:10.1093/jhered/esw039.
Mooney, C. 2014. “This Picture has Creationists Terrified.” February 4, 2014. http://www.motherjones.com/politics/2014/01/bill-nye-creationism-evolution.
Moran, L. A. 2015. “Creationists Discover that Human and Chimp Genomes are only about 70% Similar!” June 17, 2015. http://sandwalk.blogspot.com/2015/06/creationists-discover-that-human-and.html.
Quinn, J. J., and H. Y. Chang. 2016. “Unique Features of Long Non-Coding RNA Biogenesis and Function.” Nature Reviews Genetics 17 (1): 47–62. doi:10.1038/nrg.2015.10.
Riethman, H., A. Ambrosini, C. Castaneda, J. Finklestein, X. L. Hu, U. Mudunuri, S. Paul, and J. Wei. 2004. “Mapping and Initial Analysis of Human Subtelomeric Sequence Assemblies.” Genome Research 14 (1): 18–28. doi:10.1101/gr.1245004.
Rudd, M. K. 2014. “Human and Primate Subtelomeres.” In Subtelomeres. Edited by E. J. Louis and M. M. Becker, 153–164. New York: Springer Science and Business Media.
Tanaka, H., S. Abe, N. Huda, L. Tu, M. J. Beam, B. Grimes, and D. Gilley. 2012. “Telomere Fusions in Early Human Breast Carcinoma.” Proceedings of the National Academy of Sciences USA 109 (35): 14098–14103. doi:10.1073/pnas.1120062109.
Tanaka, H., M. J. Beam, and K. Caruana. 2014. “The Presence of Telomere Fusion in Sporadic Colon Cancer Independently of Disease Stage, TP53/KRAS Mutation Status, Mean Telomere Length, and Telomerase Activity.” Neoplasia 16 (10): 814–823. doi:10.1016/j.neo.2014.08.009.
Tomkins, J. 2013. “Alleged Human Chromosome 2 ‘Fusion Site’ Encodes an Active DNA Binding Domain Inside a Complex and Highly Expressed Gene—Negating Fusion.” Answers Research Journal 6: 367–375.
Tomkins, J. P., and J. Bergman. 2011. “Telomeres: Implications for Aging and Evidence for Intelligent Design.” Journal of Creation 25 (1): 86–97.
Tsipouri, V., M. G. Schueler, S. Hu, NISC Comparative Sequencing Program, A. Dutra, E. Pak, H. Riethman, and E. D. Green. 2008. “Comparative Sequence Analyses Reveal Sites of Ancestral Chromosomal Fusions in the Indian Muntjac Genome.” Genome Biology 9 (10): R155. doi:10.1186/gb-2008-9-10-r155.
Tu, L., N. Huda, B. R. Grimes, R. B. Slee, A. M. Bates, L. Cheng, and D. Gilley. 2015. “Widespread Telomere Instability in Prostatic Lesions.” Molecular Carcinogenesis 55 (5): 842–852. doi:10.1002/mc.22326.
Ventura, M., C. R. Catacchio, S. Sajjadian, L. Vives, P. H. Sudmant, T. Marques-Bonet, T. A. Graves, R. K. Wilson, and E. E. Eichler. 2012. “The Evolution of African Great Ape Subtelomeric Heterochromatin and the Fusion of Human Chromosome 2.” Genome Research 22 (6): 1036–1049. doi:10.1101/gr.136556.111.
Voronin, D. A., and E. V. Kiseleva. 2008. “Functional Role of Proteins Containing Ankyrin Repeats.” Cell and Tissue Biology 49 (12): 989–999.
Xie, C., J. Yuan, H. Li, M. Li, G. Zhao, D. Bu, W. Zhu, W. Wu, R. Chen, and Y. Zhao. 2013. “NONCODEv4: Exploring the World of Long Non-Coding RNA Genes.” Nucleic Acids Research 42: D98-103. doi:10.1093/nar/gkt1222.
Yunis, J. J., and O. Prakash. 1982. “The Origin of Man: A Chromosomal Pictorial Legacy.” Science 215 (4539): 1525–1530.